Revista Española de Derecho Europeo
88 | Octubre – Diciembre 2023
pp. 49-108
Madrid, 2023
DOI:10.37417/REDE/num88_2023_
© Marcial Pons Ediciones Jurídicas y Sociales
© José Carlos Cano Montejano
ISSN: 1579-6302
Recibido: 24/07/2023 | Aceptado: 05/09/2023
LA AGENCIA EUROPEA DE MEDICAMENTOS: ANÁLISIS JURÍDICO Y COMPARACIÓN CON LA FDA ESTADOUNIDENSE
The European Agency of Medicines: legal analysis and comparison with the American FDA
José Carlos Cano Montejano*
Resumen: El presente trabajo tiene como objeto un análisis jurídico del estatuto de la Agencia Europea de Medicamentos, desde una perspectiva centrada en las cuestiones atinentes a la aprobación de los medicamentos, los procedimientos vigentes al momento de la redacción, llevando a cabo una comparación entre los métodos y sistemas que sigue la FDA estadounidense, buscando una mejora regulatoria que se propone en las conclusiones del trabajo.
Palabras CLAVE: regulación; Agencia Europea de Medicamentos; transparencia; FDA.
Abstract: The purpose of this paper is a legal analysis of the statute of the European Medicines Agency, from a perspective focused on the issues related to the approval of drugs, the procedures in force at the time of drafting this paper, carrying out a comparison between the methods and systems followed by the US FDA, seeking a regulatory improvement in the EMA statute and protocols, that is proposed in the conclusions of the paper.
KEYWORDS: regulation; European Agency of Medicines; transparency; FDA.
SUMARIO: INTRODUCCIÓN.— 1. ESTRUCTURA JURÍDICA DE LA EMA: 1.1 Orígenes 1.2 Estatuto Jurídico 1.3 Funciones 1.4 Composición 1.5 Actuación y toma de decisiones 1.6 Regulación.— 2 EL ITER DEL MEDICAMENTO EN LA EMA: 2.1 Investigación y desarrollo 2.2 Asesoría científica 2.3 Evaluación 2.4 Autorización 2.5 Acceso 2.6 Monitarización de la seguridad.— 3. ASPECTOS PROBLEMÁTICOS: TRANSPARENCIA, CÓDIGO DE CONDUCTA Y CONFLICTOS DE INTERÉS: 3.1 Publicidad de la información y acceso a documentos 3.2 Códigos de Conducta 3.3 Gestión de conflictos de interés.— 4. COMPARACIÓN METODOLÍGICA CON LA FDA: 4.1 Creación y evolución. 4.2 Organización y estructura 4.3 Resultados comparativos.— 5. PROPUESTAS DE MEJORA REGULATORIA: 5.1 Criterios para la elección de los miembros de los órganos 5.2 Separación de las funciones de asesoría científica y emisión de opinión 5.3 Clarificación y simplificación de la estructura orgánica 5.4 Reforma del actual enfoque formalista de la transparencia 5.5. Refuerzo del derecho de defensa 5.6 Agilización de los trámites.— CONCLUSIÓN
INTRODUCCIÓN
En este trabajo se pretende afrontar un reto importante, en la medida en que se quiere analizar una cuestión que ha sido de una actualidad inequívoca, ya que la pandemia nos ha confrontado con algunas cuestiones que para la mayoría de la ciudadanía eran perfectamente desconocidas, bastante inasequibles, y reservadas única y exclusivamente a expertos, empresas farmacéuticas, profesionales del sector de la salud, y demás categorías de implicados en el sector sanitario, es decir, a aquellos que participan cotidianamente en la tarea transcendental de preservar y mejorar la salud y las condiciones de vida.
En este sentido, la crisis provocada por la COVID ha despertado en la mayoría de nosotros inquietudes que nunca antes se nos habían planteado o suscitado: nos ha enfrentado a cuestiones que hasta hace no demasiado tiempo eran exorbitantes, y excedían del conocimiento medio del ciudadano de a pie; sin embargo, el iter de los tratamientos —y especialmente de la vacunación frente a la pandemia— la autorización de medicamentos para luchar contra sus devastadores efectos, han puesto en la picota instituciones y organismos que con anterioridad sólo eran conocidos tangencialmente o de manera periférica —incluso para el público especializado—: en concreto, nos referimos a la Agencia Europea de Medicamentos (en adelante, “EMA” o “la Agencia”).
Como hemos indicado, la EMA ha sido determinante en relación con numerosos hitos de la pandemia —métodos de tratamiento, sistemas de protección, autorización de medicamentos, y finalmente, aprobación de las diversas vacunas relativas al virus causante de la COVID en sus diferentes manifestaciones y mutaciones—. Pero la EMA tiene una transcendencia que va mucho más allá de estas puntuales y coyunturales circunstancias —que ya casi han alcanzado la categoría de pretéritas, tan fútil es la memoria humana—: la realidad es que la Agencia Europea del Medicamento es un organismo de una importancia y entidad muy relevantes, en la medida en que articula, da forma y expresa la competencia que las Instituciones Europeas despliegan en materia de sanidad y de protección de la salud: estas atribuciones son realmente singulares en el conjunto de las competencias que los Tratados europeos las atribuyen, en la medida en que la salud pública y su protección se enmarca en dos categorías distintas de competencias, algo que realmente las particulariza y significa respecto al resto de las que se despliegan por la UE a través de esas mismas Instituciones.
El Tratado de Funcionamiento de la UE nos indica que las Instituciones Europeas ostentan una competencia compartida con los Estados miembros en relación con los asuntos comunes de seguridad en materia de salud pública 1, de esta forma, se encuadra dentro de las categorías competenciales en las cuales concurren dos regulaciones: la nacional y la de la Unión, interactuando entre ambas, pero partiendo de dos principios moduladores de la actividad de las Instituciones Europeas: el principio de subsidiariedad y el de proporcionalidad.
La realidad es que actualmente gran parte de la regulación que procede de las Instituciones Europeas se articula a través de la realización de competencias compartidas, en las que se manifiestan de manera particularmente palmaria los principios de primacía y de eficacia directa del ordenamiento jurídico que se expresa en el Derecho de la Unión 2. Esta cohabitación entre regulaciones —la nacional y la europea— parte de un principio unívoco: los Estados miembros de la UE tienen derecho a regular la materia en cuestión —en este caso, aquellas relacionadas con la protección de la salud—, pero si la UE desarrolla aspectos ya tratados por la legislación nacional, ésta deberá acomodarse a los imperativos emanados desde Bruselas. De esta forma, la convivencia entre ambos ordenamientos es relativamente inestable, ya que el Derecho nacional debe ceder y amoldarse a los principios que se le impongan y establezcan desde la regulación europea.
Además, como hemos señalado, el propio Tratado de Funcionamiento de la Unión Europea (en adelante “TFUE”) establece que también es competencia de las Instituciones Europeas la protección y mejora de la salud humana 3, aunque esta atribución se plasma como competencia de apoyo y coordinación, es decir, en un plano inferior a las compartidas, que pueden conllevar un carácter excluyente respecto del Derecho interno.
Conviene indicar todas estas cuestiones al inicio de este trabajo ya que, por un lado, permiten intuir la complejidad de la relación que se suscita entre el Derecho interno y del Derecho de la UE en el ámbito de la salud, pero también —desde otra perspectiva— sirve para considerar en su justa medida la importancia que las cuestiones relacionadas con la salud y su protección adquieren en el marco de la actividad de las Instituciones Europeas, siendo una de sus concreciones más importantes la Agencia Europea de Medicamentos.
Se trata éste de un organismo dotado de unos caracteres que la significan desde una perspectiva comparativa a nivel internacional, no sólo por sus atribuciones y la trascendencia de sus decisiones a nivel europeo, sino por la forma en que se manifiestan estas potestades, que hacen que sea diferente a otras instituciones de carácter equivalente y con competencias equiparables, como puede ser la Food and Drug Administration en Estados Unidos.
Por esta razón, el esfuerzo más importante de este trabajo se va a manifestar en el análisis de los procedimientos de aprobación de medicamentos a nivel europeo, en el estudio de diversos elementos atinentes a la transparencia en su labor, y también vamos a centrarnos en ofrecer algunos elementos comparativos con la Food and Drug Administration (en adelante, “FDA”), que puedan servir para contrastar y cotejar algunas perspectivas respecto de ambas agencias, expresando así posibles líneas de mejora en la actual regulación vigente respecto de la EMA.
De este modo, dedicaremos un capítulo a la explicación del itinerario que sigue un medicamento a nivel europeo para que pueda ser aprobado y, eventualmente, pasar a disposición del paciente. En este sentido, aprovecharemos algunos elementos para manifestar cuestiones que —en nuestra opinión— afectan a la transparencia y a la difícil y trascendente cuestión de los conflictos de interés.
Es evidente que, a lo largo del trabajo, será necesario citar y ofrecer referencias de algunos de los conflictos que se han suscitado en el marco de decisiones emitidas por la EMA, y en los que particular y explícitamente se ponen de manifiesto algunos de los elementos que queremos resaltar: no se trata de llevar a cabo una exposición aséptica, nuestro objetivo es ejemplificar con casos reales aquellos elementos que se nos antojan especialmente complejos, y a los que convendría dar una respuesta, no sólo mejorando los mecanismos procedimentales y de trabajo, sino también e incluso como auténticas propuestas de lege ferenda, es decir, que eventualmente pudiesen asumirse en reformas legislativas respecto del marco normativo vigente.
Por último, propondremos un elenco de posibles medidas que pudiesen contribuir a una mejora del ambiente regulatorio en el ámbito de la Agencia, cuya labor —no podemos obviarlo— permea todos los aspectos relativos a la protección de salud para el conjunto de la UE, siendo que manifiesta, encarna y expresa un servicio público indiscutible por su propia naturaleza, como es la protección de la salud como realización y expresión del bien común y del interés general.
1. ESTRUCTURA DE LA EMA
1.1. Orígenes
Como hemos indicado, la Agencia Europea de Medicamentos comienza su funcionamiento el 26 de enero de 1995, aunque su estatuto básico se encuentra precisado en una norma que tiene una datación claramente posterior —el Reglamento 726/2004 4—, que establecía un Título específicamente dedicado a especificar las responsabilidades y la estructura administrativa de la Agencia; pero conviene partir de una premisa antes de seguir adelante con la exposición de la legislación a la que se somete este ente descentralizado: en el sitio web de la EMA 5 hemos localizado más de quinientos documentos que tienen relación con la regulación aplicable a la misma —y esto restringiendo la búsqueda exclusivamente a aquellos que tienen relación con la salud humana— 6, si extendiésemos a otras categorías los criterios de búsqueda (medicamentos de uso veterinario por ejemplo), se pueden encontrar alrededor de 1.800 documentos que tienen relación con la regulación y normativa aplicable.
A esta cuestión se hará referencia con posterioridad, pero conviene referirla en estos momentos, porque permite ponderar con cierta perspectiva la magnitud de la normativa existente alrededor de la EMA, y la trascendencia y amplitud que alcanza la misma. También permite atisbar prima facie la gran complejidad de los procedimientos y mecanismos en los que la Agencia desarrolla sus potestades.
La Unión Europea cuenta con 23 agencias descentralizadas 7, entre las que se encuentra la EMA 8 —por citar las más conocidas podemos referir: la EBA (Autoridad Bancaria Europea), la EASA (Agencia Europea de Seguridad Aérea), la ECHA (Agencia Europea de Sustancias y Mezclas Químicas), la EEA (Agencia Europea del Medioambiente), la Agencia Europea del Control de la Pesca (EFCA), la Autoridad Europea de Seguridad Alimentaria (EFSA)—; por su parte, en España tienen su sede tres Agencias: la Oficina de Propiedad Intelectual de la Unión Europea (EUIPO), con sede en Alicante, en Bilbao se encuentra la Agencia Europea para la Seguridad y Salud en el Trabajo (OSHA) y en Vigo la Agencia Europea de Control de la Pesca (EFCA).
En total, a nivel de la UE, nos encontramos con 72 órganos, agencias —ejecutivas y descentralizadas 9— e Instituciones Europeas que se reparten por todos los Estados miembros; la EMA en concreto tiene su sede en los Países Bajos —en la ciudad de Ámsterdam 10—; el estatus de agencia descentralizada implica una serie de elementos que manifiestan cierta independencia, pero jerárquica y funcionalmente se encuentra sujeta a la Comisión Europea, que propone a su Director Ejecutivo y que, a su vez, es nombrado por el Consejo de Administración de la Agencia, después de haber presentado y defendido su candidatura ante el Parlamento Europeo 11.
1.2. Estatuto jurídico
La naturaleza jurídica de la Agencia Europea de Medicamentos es ciertamente singular, evidentemente es un órgano sometido al Derecho Público de la Unión, es decir, estamos ante un ente de naturaleza administrativa 12 —que encuentra parangón en los Estados miembros en las correspondientes agencias nacionales—, y, por lo tanto, se somete a todos aquellos elementos que integran el corpus jurídico que en el ámbito comunitario resulta aplicable no sólo al estatuto de sus miembros y empleados, sino también a los principios regulatorios que rigen la actuación de los distintos organismos e instituciones europeos, y que se puede calificar de auténtico Derecho Administrativo Europeo 13.
En este sentido, ya en el año 2001, el Servicio Jurídico de la Comisión Europea definía las agencias europeas como
órganos descentralizados con las siguientes características: creación por Reglamento; personalidad jurídica; órganos de gestión autónomos; independencia financiera; personal cubierto por los Reglamentos de funcionarios y por las Condiciones para el empleo de otros funcionarios de las Comunidades Europeas; y objetivos y encargos específicos 14.
La Unión Europea nos ofrece también una definición propia de agencia, al establecer que es
un organismo de Derecho público europeo, distinto de las Instituciones Comunitarias (Consejo, Parlamento, Comisión, etc.) y que posee una personalidad jurídica propia. Las agencias comunitarias se crean mediante un acto comunitario de Derecho derivado con el fin de desempeñar una tarea específica de naturaleza técnica, científica o de gestión que se especifica en el correspondiente acto fundacional 15.
Por su parte, la Comisión Europea entiende que una agencia reguladora es:
una entidad jurídica autónoma creada por la autoridad legislativa para participar en la regulación de un sector a escala europea y en la ejecución de una política comunitaria determinada 16
Es indudable que la EMA es una de las agencias emblemáticas en la UE, participa de todos los elementos que se han invocado con anterioridad, y —por lo tanto— tiene esa naturaleza jurídica que se engloba bajo el paraguas del Derecho Público Europeo 17, con carácter autónomo, y cuya actuación se refiere a la regulación de un sector concreto a escala europea.
Es también importante señalar que las Agencias europeas, para su creación a través de una disposición jurídica de Derecho Europeo derivado —como hemos visto en el caso de la EMA, se recurrió a un Reglamento— tienen que invocar una base jurídica efectiva en los Tratados Europeos. De hecho, normalmente se invocó para la creación de la mayoría de las agencias descentralizadas el artículo 308 del TUE 18, que tradicionalmente se utilizó para numerosas acciones comunitarias, y que implica el consenso y unanimidad en el Consejo para que pudiesen salir adelante 19.
Sin embargo, para la EMA se recurrió a un artículo distinto del TCE para su creación: el artículo 175.1 20 —que estaba originalmente ideado para llevar a cabo acciones comunes respecto del medio ambiente a nivel europeo—, ya que —por una parte— suponía una mayor implicación del Parlamento Europeo, y de otra bastaba con la mayoría cualificada en el Consejo 21.
Estas cuestiones pueden parecer periféricas o tangenciales ya que sólo hacen referencia a la naturaleza jurídica de la EMA, sin embargo, son de una trascendencia capital, porque nos indican las imperiosas necesidades de consenso que ya se sintieron en los albores de su creación, y que —por lo tanto— debería permanecer en el espíritu de las acciones y decisiones realzadas y tomadas por la misma, ya que afectan de forma transversal y radical al conjunto de los países miembros de la UE, y a toda la ciudadanía europea. Pero es evidente que el consenso también implica una serie de servidumbres y compromisos que podrían ir más allá y exceder los parámetros derivados exclusivamente de la pura evidencia científica, al fundamentarse en criterios de distinta naturaleza. Este hecho será puesto en evidencia en lo sucesivo en repetidas ocasiones.
1.3. Funciones
El acto jurídico fundacional de la EMA, el Reglamento 726/2004, nos explicita las misiones que deberá cumplir la misma:
La Agencia tendrá por misión proporcionar a los Estados miembros y a las instituciones de la Comunidad el mejor asesoramiento científico posible sobre cualquier cuestión relacionada con la evaluación de la calidad, la seguridad y la eficacia de medicamentos de uso humano o veterinario que se le someta, de conformidad con lo dispuesto en la legislación comunitaria sobre medicamentos 22.
Esta genérica declaración nos recuerda inequívocamente la naturaleza de la competencia que se realiza a nivel europeo desde la Agencia Europea de Medicamentos, ya que su acción debe basarse en esa naturaleza compartida y de apoyo y coordinación de la atribución recibida, para así poder prestar a los Estados miembros y a las Instituciones europeas un asesoramiento científico independiente y solvente, que para ser calificado como tal debería estar libre de influencias, criterios, parámetros o elementos que pudiesen eventualmente desnaturalizar el objetivo fijado en su misión.
Por otra parte, ese asesoramiento científico se plasma entre otros en:
— La coordinación de la evaluación científica de la calidad, seguridad y eficacia de los medicamentos cuya comercialización esté sujeta a procedimientos comunitarios de autorización;
— La puesta a disposición del público de informes de evaluación, resúmenes de características de productos, etiquetados y prospectos de los medicamentos;
— El control de los medicamentos autorizados en la Unión, facilitando asesoramiento sobre las medidas necesarias para garantizar el empleo eficaz y seguro de dichos medicamentos, en particular mediante la evaluación y coordinación de la aplicación de las obligaciones de farmacovigilancia;
— Garantizar la difusión de información sobre las reacciones adversas de los medicamentos autorizados en la Unión, por medio de una base de datos que los Estados miembros podrán consultar de forma permanente; los profesionales de los servicios sanitarios, los titulares de autorizaciones de comercialización y el público en general tendrán derecho de acceso a las bases de datos, jerarquizados según los casos; estas bases de datos garantizarán la protección de los datos personales;
— Asesorar a las empresas sobre la realización de las diferentes pruebas y estudios necesarios para demostrar la calidad, la seguridad y la eficacia de los medicamentos;
— Verificar que se respetan las condiciones impuestas por la legislación comunitaria relativa a los medicamentos y por las autorizaciones de comercialización de medicamentos autorizados por la EMA;
Sólo hemos enumerado algunas de las funciones encomendadas por la EMA centrándonos en las que hacen referencia a los medicamentos dirigidos al consumo humano y a su comercialización —recordemos que también lleva a cabo importantes labores en el ámbito veterinario—; de hecho, se puede afirmar que la aprobación de la comercialización de medicamentos y tratamientos a nivel europeo es la más importante de las actividades asignadas a esta agencia, y que se manifiesta en el primer apartado expresado con anterioridad, relativo a los medicamentos que estén sometidos a procedimientos comunitarios de autorización.
1.4. Composición
En otro orden de cosas, el artículo 56 del Reglamento 726/2004 nos enumera los órganos que componen la Agencia Europea de Medicamentos:
— El Director Ejecutivo, propuesto por la Comisión y nombrado por el Consejo de Administración después de someterse a un hearing —una sesión de control— en el Parlamento Europeo. Su mandato es por cinco años, pudiendo renovarse por uno suplementario.
— El Consejo de Administración, que —como establece el artículo 65.1 del Reglamento 23— está compuesto por representantes de los Estados miembros, por dos representantes de la Comisión y dos del Parlamento Europeo. Además, forman parte del mismo otros representantes de organizaciones de médicos, pacientes y veterinarios —propuestos por la Comisión Europea—. La decisión final respecto al nombramiento de estos miembros recae en el Consejo.
— El Comité de Medicamentos de Uso Humano
— El Comité de Medicamentos de Uso veterinario
— El Comité de Medicamentos huérfanos
— El Comité de Medicamentos a base de plantas
— Una Secretaría que presta asistencia técnica, científica y administrativa a los Comités.
Respecto a este somero organigrama es necesario aclarar algunas cuestiones relevantes, en la medida en que afectan al fondo del presente trabajo. El Consejo de Administración —como hemos indicado— está formado eminentemente por representantes de los Estados miembros, ya que los miembros de las organizaciones profesionales y de pacientes también se aprueban por el propio Consejo; de esta forma, resulta oportuno preguntarse por los criterios que deben concurrir en los candidatos que se proponen al Consejo de Administración desde los Estados miembros, siendo que el texto del Reglamento —artículo 65.2 24— siendo parco, nos permite entrever que los baremos no tienen por qué estar exclusivamente relacionados con su especialización en cuestiones médicas o veterinarias, es decir, criterios que concurran en la formación y currículo del candidato, bastando que el candidato tenga conocimientos pertinentes en materia de gestión, y sólo si conviniese, en otras materias especializadas del ámbito sanitario.
Es evidente que el criterio de proposición de miembros del Consejo de Administración de la Agencia Europea de Medicamentos queda muy abierto, pudiendo ser permeado por consideraciones periféricas a las exclusivamente sanitarias, como pudiesen ser eventualmente criterios de adscripción política o de afinidad gubernamental. Al mismo tiempo, no se puede perder de vista que las decisiones en el seno del Consejo de Administración se toman por mayoría de dos tercios, es decir, por una mayoría cualificada 25, en la cual —al igual que ocurre en el funcionamiento del Consejo como órgano central en el esquema institucional de la Unión Europea— las tendencias y las posiciones de determinados Estados pueden ser determinantes en la orientación del voto de otros países en su ámbito de influencia, algo que también puede reflejarse en la actuación del Consejo de Administración de la EMA.
En este sentido, es importante remarcar que el Consejo de Administración —por ejemplo— puede invitar a sus sesiones a los presidentes de los Comités científicos, que no tendrán voto en la decisión final adoptada 26; es decir, las medidas que se adopten por el Consejo sólo quedan permeadas por la opinión científica expresada en los Comités en la medida en que se asuman por sus miembros, que —como hemos indicado— pueden tener consideraciones exorbitantes a la mera evidencia científica 27.
De esta forma, es importante resalar que los Comités científicos son los que verdaderamente articulan el trabajo de la EMA, en la medida en que tanto en el Comité de medicamentos de uso humano como en el Comité de medicamentos de uso veterinario reposa la responsabilidad de emanar los dictámenes e informes relativos a los medicamentos que se estén sometiendo a su consideración.
Los miembros de los Comités —y sus suplentes— son propuestos por los Estados miembros previa consulta al Consejo de Administración de la Agencia, pudiendo ser elegidos por su función y experiencia en la evaluación de medicamentos de uso humano o veterinario 28. Asimismo, cada Comité podrá nombrar por cooptación otros cinco miembros adicionales por razón de su especialización científica —pero siempre dentro de una lista de expertos científicos elaborada por los Estados miembros y la Agencia—. A las reuniones de los Comités —o de sus grupos de trabajo— pueden asistir tanto el Director Ejecutivo como los representantes de la Comisión en el Consejo de Administración.
Es interesante el matiz que introduce el artículo 61.6 29, ya que parte de la premisa de la colaboración con los organismos nacionales de autorización de comercialización, de forma que los miembros de los comités y los expertos que estén trabajando en la evaluación de algún medicamento deberían basarse en la experiencia y recursos científicos de aquellos. Además, a la agencia nacional correspondiente se le encarga la labor de supervisar la independencia de la evaluación que se haya llevado a cabo desde la EMA y sus comités. Explícitamente se expresa la obligación que incumbe a los Estados miembros de abstenerse de imponer a los miembros de comités y expertos cualquier instrucción incompatible con sus funciones dentro de la Agencia.
Hacemos particular hincapié en nuestro análisis en la ponderación y estudio de los elementos que hacen referencia a la independencia, imparcialidad y transparencia en los procedimientos atinentes a las actividades de la Agencia, en la medida en que —como iremos poniendo de manifiesto a lo largo del trabajo— es aquí donde entendemos se puede llevar a cabo una contundente labor de mejora, necesaria para prestigiar la labor de la misma, y también para garantizar y asegurar que todas las partes implicadas puedan disponer de la máxima información y puntos de referencia que permitan enjuiciar la adecuación y razonabilidad de las diferentes decisiones que tome, sea en el ámbito de la aprobación de comercialización de medicamentos, o en el plano de tratamientos preventivos y de vacunación.
1.5. Actuación y toma de decisiones
Los Comités para la toma de decisiones se basan en una interesante noción que se manifiesta y resume en el logro del consenso científico 30, de manera que en su defecto se recurriría al voto por mayoría para alcanzar la decisión correspondiente. Es importante recordar que los miembros del Comité representan a las agencias nacionales de cada Estado miembro. Es necesario exponer, aunque sea someramente, el funcionamiento interno de los Comités para poder entender el contenido de este concepto jurídico indeterminado que se refleja el consenso científico indicado.
Dentro de los Comités se crean —conforme a las reglas expresadas en sus propios reglamentos internos— grupos de trabajo y grupos científicos consultivos, que son los órganos en los que se articula de manera efectiva el trabajo de los Comités. Cada Comité a su vez designa un ponente y un ponente adjunto —rapporteur y co-rapporteur, cuando se trate de evaluar la aprobación de un medicamento—, que elaborarán un proyecto de informe de evaluación, cuya sustancia y elementos básicos relevantes se publicarán conforme a los criterios expresados en el propio Reglamento —artículos 38.3 y 13.3 31—: es importante señalar que se exige explícitamente que ese informe esté motivado, y —por lo tanto— parte de ese componente esencial a publicar debe residir necesariamente en la motivación que se haya acompañado al informe. Este documento recibe el nombre de Informe público Europeo de Evaluación —European Public Assessment Report (EPAR)—. Es interesante resaltar que el solicitante de la autorización podrá pedir una revisión del examen llevado a cabo en el dictamen por el ponente o co-ponente, que se llevará a cabo por expertos distintos de los que evaluaron en primer término, y sólo respecto de aquellas cuestiones que el solicitante haya entendido sean susceptibles de aclaración, partiendo de que se utilizará únicamente la evidencia científica disponible en el momento de la elaboración del primer dictamen 32.
También conviene señalar en estos momentos que, normalmente, y en la práctica casi sin excepciones, el proyecto de informe de evaluación elaborado por el ponente o su suplente se siguen por la Agencia en su decisión final, asumiendo sus posiciones y, por lo tanto, avalando el trabajo y la labor previa e independiente que llevan a cabo estos expertos, que —por otra parte— suelen disponer de todos los elementos y parámetros de valoración necesarios para emitir un informe fundamentado y contrastado 33. Si la Agencia se apartase del proyecto de informe, debería basarse en circunstancias conocidas y motivadas contundentemente, dado lo excepcional y exorbitante de la situación, además es una obligación que deriva de la legislación aplicable vigente 34.
1.6. Regulación
De forma congruente, el Reglamento que sienta las bases del funcionamiento de la Agencia hace un especial énfasis en la independencia de todos los miembros de la misma —ya sean de Comités, del Consejo de Administración, de Grupos de trabajo o de los asesores científicos contratados a través del sistema de manifestaciones de interés—. El Código de Conducta de la Agencia deberá regular especialmente las situaciones de conflictos de interés, siempre poniendo el énfasis en la búsqueda de la salvaguarda del interés general, de forma que esas circunstancias puedan ser públicas y accesibles. 35 También les resulta de aplicación la normativa vigente respecto de la Comisión Europea en esta materia, y que se plasma en el Código Europeo de Buena Conducta Administrativa, al quedar equiparados los miembros y staff de la EMA al personal de esta Institución Europea 36.
El Comité de Medicamentos de Uso Humano cuenta desde 2007 con un Reglamento interno de procedimiento 37 en el que se regula de manera exhaustiva la relación que el ponente y el co-ponente pueden tener antes y durante la fase de evaluación de un medicamento con las partes interesadas, y especialmente con aquellos que han solicitado la aprobación del mismo. De esta forma, cuando se entiende que es conveniente el nombramiento de un co-rapporteur su labor se dirige a la elaboración de un informe crítico paralelo del que haya emitido el ponente principal, e incluso se le puede exigir la elaboración de un informe completo complementario; además ambos informes pueden ser sometidos a la revisión o peer review llevada a cabo por expertos miembros del Comité o de alguno de los Grupos asesores científicos. 38 En ese mismo reglamento se fijan las normas que deben regir las votaciones, partiendo de la premisa de que se debe conseguir el consenso en la medida de lo posible —es decir, la unanimidad—, en otro caso se tendría que decidir por mayoría de dos tercios, o por mayoría absoluta de los miembros del Comité, dependiendo de las circunstancias.
En el mismo sentido, se recoge en el citado reglamento que las opiniones divergentes deberán hacerse públicas, constando por escrito y expresando los fundamentos para su oposición. Al mismo tiempo, se afirma que, en caso de juzgarse oportuno, las actas y minutas del Comité deberán también hacerse públicas 39.
Recordemos que no todos los medicamentos tienen que solicitar su aprobación a través de la Agencia Europea de Medicamentos, aunque sí todos aquellos que explícitamente quedan recogidos en el Anexo del Reglamento 726/2004, y que hacen referencia a aquellos que hayan sido desarrollados a través de procesos biotecnológicos determinados, y en concreto —respecto a aquellos destinados al uso humano— los que contengan una nueva sustancia activa para el tratamiento del síndrome de inmunodeficiencia adquirida, del cáncer, de los trastornos neurodegenerativos y de la diabetes, todo ello con efectos a partir de 20 de mayo de 2008. También han quedado incluidas en este elenco las enfermedades autoinmunes y otras disfunciones inmunes, así como las enfermedades víricas. De esta forma podemos observar la trascendencia, importancia y alcance de los medicamentos que necesitan contar con la aprobación de la Agencia para poder ser comercializados legalmente en los Estados miembros de la UE 40, sin tomar en consideración el hecho imponderable de que esta autorización tiene una radical significación respecto del resto de los países de la comunidad internacional, porque constituye un precedente inequívoco y contundente de la eficacia y conveniencia para permitir también su comercialización en sus respectivos mercados y jurisdicciones 41.
También son relevantes respecto al marco jurídico aplicable a la Agencia Europea de Medicamentos una serie de normas de Derecho derivado de la UE, que se manifiestan en varias Directivas que discriminan entre los productos dedicados al uso humano y los que se dirigen al uso veterinario. En concreto, hay que citar la Directiva 2001/83/EC, relativa al establecimiento de un código comunitario relativo a los medicamentos de uso humano, y que ha sido sometida a diversas modificaciones a lo largo de su existencia. 42
En lo que hace referencia a la legislación relativa a la farmacovigilancia conviene citar el Reglamento Nº. 1235/2010, y el Reglamento 1027/2012, la Directiva 2010/84/EU y la Directiva 2012/26/EU. También hay un acto de ejecución, que se plasma en el Reglamento de Ejecución Nº 520/2012. En relación con los medicamentos relativos a terapias avanzadas es necesario citar el Reglamento 1394/2007.
Es evidente que el objeto de este trabajo no es la exposición completa y omnicomprensiva de todas las facetas regulatorias relativas a la Agencia Europea de Medicamentos, pero sí hemos intentado centrarnos en aquellos elementos relativos a su funcionamiento interno que van a servir de base a los elementos que se van a ir poniendo de manifiesto a lo largo de las páginas siguientes, de manera que podamos tener una visión de conjunto que sirva para poner de manifiesto la trascendencia que hay que otorgar a la articulación de la transparencia en esta organización, por la relevancia que adquiere la misma, y por la importancia de que sus métodos, procedimientos y mecanismos estén influidos e imbuidos de una auténtica cultura relativa a este principio irrenunciable en cualquier organismo público, sea de naturaleza nacional o europeo.
2. EL CAMINO DE UN MEDICAMENTO EN LA EMA
Antes de nada, conviene comenzar diciendo que la EMA forma parte esencial del Sistema Regulatorio de medicamentos para la UE 43, que engloba a 30 países —los 27 miembros de la UE, Islandia, Liechtenstein y Noruega—, de la que forman parte también 31 agencias nacionales, de este modo forma un network que coopera mutuamente y comparte expertise y experiencia respecto a la regulación de los medicamentos para prácticamente todo el territorio europeo.
El elemento determinante y básico es la autorización de comercialización de medicamentos que, en Europa, se puede llevar a cabo a través de tres mecanismos distintos:
— El procedimiento centralizado, que permite comercializar un medicamento para todo el espacio de la UE a través de la EMA, agencia que asesora en todo el procedimiento y propone una resolución respecto a la solicitud presentada a la Comisión Europea, que es quien se encarga de decidir de forma definitiva si se autoriza o se deniega la solicitud. Esta autorización es válida en todos los países de la UE, y además se prevalece del mercado único europeo que se aplica también a este tipo de productos 44. Como veremos, para determinados medicamentos es obligatorio recurrir a la aprobación de la EMA, pero para la gran mayoría no es necesario ya que basta con la autorización de las agencias nacionales correspondientes.
— El procedimiento descentralizado, que es aquel según el cual las compañías pueden solicitar la autorización simultánea de un medicamento en más de un Estado miembro de la UE, siempre que todavía no haya sido aprobado en ninguno de ellos, y no haya obligación de recurrir al procedimiento centralizado por la naturaleza del mismo.
— El procedimiento de reconocimiento mutuo, que tiene lugar cuando una empresa tiene una autorización de comercialización para un Estado miembro y solicita a otras agencias nacionales que se reconozca la aprobación que ha obtenido en ese país.
Vamos únicamente a ocuparnos del iter que sigue la solicitud de aprobación de un medicamento ante la Agencia Europea de Medicamentos, para poder así mejor entender con posterioridad algunos de los elementos que se irán poniendo de manifiesto a lo largo de este trabajo, sobre todo en relación con la FDA americana. Se puede resumir el camino de la solicitud en la EMA en seis pasos que se pueden desglosar en: investigación y desarrollo, asesoría científica, evaluación, autorización, acceso y monitorización de la seguridad; vamos a contemplarlos de manera singularizada:
2.1. Investigación y desarrollo
La cantidad de sustancias que se estudian cada año en relación con nuevos medicamentos es de varios millares, fundamentalmente su desarrollo se lleva a cabo a través de compañías farmacéuticas y biotecnológicas de tamaño muy distinto; es evidente que de todas ellas sólo una pequeña cantidad pasará a la fase de ensayos clínicos, y de estas sólo una fracción ínfima recibirá la aprobación definitiva.
En cualquier caso, el esfuerzo de investigación y financiero que se lleva a cabo es muy importante, y se realiza dentro de unos parámetros que se salvaguardan por la EMA conforme a baremos internacionales, con independencia de que el desarrollo se esté llevando a cabo fuera o dentro de la UE. Esta labor de vigilancia se extiende hasta la supervisión de la producción científica en la materia 45. De este modo, los medicamentos se prueban primero en el laboratorio, pasando después a las pruebas clínicas —clinical trials— para poder evaluar sus efectos sobre los seres humanos y sus eventuales efectos colaterales. Los ensayos clínicos con pacientes se autorizan por las agencias nacionales competentes, no por la EMA.
En todo caso, las normas que rigen los ensayos clínicos y la investigación se denominan good clinical practice 46, y se aplican al diseño de los estudios que se lleven a cabo, a la presentación de los resultados, para que se puedan considerar científicamente solventes, también desde una perspectiva ética. En este sentido, conviene indicar que la EMA no alienta el desarrollo de determinados medicamentos en concreto, aunque sí publica áreas en las que recomienda que se lleve a cabo una investigación. Tampoco puede ayudar financieramente a los desarrolladores porque afectaría a su posición de neutralidad y de independencia respecto al resultado final.
2.2. Asesoría científica
La EMA asiste a las empresas que estén desarrollando un medicamento en relación con los métodos y técnicas para poder demostrar la eficacia, seguridad y validez de un posible medicamento, esto se denomina scientific advice; esta evidencia se recoge y presenta a la EMA en el momento de la presentación de los resultados en relación con una solicitud de autorización. Hay diversas razones para que la Agencia se implique en esta asesoría científica: los estudios mejor diseñados y planteados tienen más posibilidades de alcanzar mejores resultados; los pacientes y enfermos podrán disfrutar de medicamentos de mayor calidad, y se les evitará que participen en ensayos clínicos con pocas perspectivas.
Durante el proceso de asesoría científica el desarrollador tiene que responder a cuestionarios que le plantea la Agencia, y ésta en ningún caso entra en una valoración de los resultados, o en una ponderación de riesgos y ventajas: las preguntas que se plantean están relacionadas con aspectos cualitativos: fabricación, cuestiones químicas, pruebas farmacéuticas y biológicas, aspectos no clínicos —pruebas toxicológicas y farmacológicas, para comprobar el comportamiento del compuesto en el laboratorio—, aspectos clínicos —resultados en pacientes voluntarios, entre otros— y cuestiones metodológicas —estudios estadísticos, análisis de datos, modelling y simulación— 47.
Dentro de la EMA, el órgano encargado de llevar a cabo el scientific advise 48 es el Comité de Medicamentos para Uso Humano —CHMP— que delega en el Grupo de Trabajo que se encarga de llevarla a cabo, el SAWP —Scientific Advice Working Party—. Este Grupo está formado por 36 miembros elegidos entre reguladores médicos de dentro de la UE, expertos del mundo académico 49, y de otros grupos de la EMA —terapias avanzadas, medicamentos infantiles, farmacovigilancia y valoración del riesgo—. Los sectores que demandan la asistencia científica están centrados en cardiología, oncología, diabetes, desórdenes neurodegenerativos y enfermedades infeccionas.
La cuestión que siempre se plantea es por qué el regulador tiene que implicarse en el desarrollo clínico y científico de un futuro medicamento: aquí se alegan razones relacionadas con la calidad del expertise de los miembros de la EMA, su obligación de compartir sus conocimientos, el desarrollo de directrices científicas que puedan servir de guía a una pluralidad de desarrolladores. Además, esta asesoría implica el cobro de unas tasas que ingresa la Agencia, y que están predeterminadas en la normativa europea vigente 50. De esta forma, el presupuesto de la EMA es de 417,5 millones de euros para 2022, de los cuales un 86 por cien corresponde a tasas y derechos pagados por los solicitantes, y un 13 por cien proviene de asignaciones directas integradas en el presupuesto comunitario.
Esta situación singulariza particularmente a la EMA dentro del panorama institucional de la UE, porque es el único organismo y agencia europea que prácticamente se autofinancia, siendo la subvención de la UE residual respecto de la cuantía de sus ingresos propios, que, en gran parte, proceden de la asistencia técnica.
Las estadísticas, en cualquier caso, afirman que el desarrollador que haya solicitado asesoría científica de la EMA tiene mayores probabilidades de recibir una ulterior aprobación que aquellos que prescindieron de la misma 51.
Al mismo tiempo, en esta fase ante la Agencia es donde mayores cuestiones se suscitan en relación con los conflictos de interés y su gestión: los miembros asesores del Grupo de Trabajo tienen que presentar una declaración de intereses que recibe una valoración de riesgo, de manera que el experto eventualmente puede ser excluido del voto, o de determinadas reuniones que traten ciertas materias.
Por otra parte, tampoco se pueden hacer públicos los resultados de la asesoría científica —al menos hasta que no se haya producido una decisión final sobre la autorización de comercialización—, de esta forma se busca salvaguardar los esfuerzos de los promotores del medicamento, que implican considerables inversiones en I+D+i. En total, el procedimiento de asesoría científica de la EMA se puede resumir en ocho pasos:
— Solicitud del desarrollador, con un briefing document.
— La Agencia le remite un cuestionario con cuestiones específicas de naturaleza científica relacionadas con la investigación que esté llevando a cabo.
— Se nombran en el SAWP —Grupo de Trabajo— dos coordinadores especializados.
— Cada coordinador forma un grupo de trabajo con expertos plurinacionales, para elaborar un informe que será discutido en el seno del SAWP.
— Reunión del SAWP con el desarrollador para discutir elementos del informe.
— Se procede a llevar a una amplia consulta a otros Comités de la Agencia, y también a expertos externos.
— Consulta a enfermos y pacientes.
— Se responde al cuestionario por el SAWP, y el CHMP emite un informe final que se remite al desarrollador.
2.3. Evaluación
Con carácter previo a la valoración de la solicitud, la Agencia comprueba si cumple con los requisitos legales y regulatorios exigibles; de este modo el solicitante tiene que proporcionar los datos y la información relevante que permitan determinar la eficacia, la calidad, la seguridad y la idoneidad del medicamento para su utilización clínica. De aquí que se celebren una serie de reuniones con representantes de la Agencia con carácter previo a la presentación de la solicitud: además en estas se deben expresar en profundidad los aspectos relacionados con el cumplimiento de la good laboratory practice, la good clinical practice, y la good manufacturing practice —que, respectivamente, hacen referencia a los ensayos clínicos y de laboratorio, y a las técnicas de producción empleadas—.
A su vez, en este capítulo es muy relevante la consideración que se hace respecto de la seguridad y la gestión de riesgos —Risk Management Plan (RMP)— que serán valorados por un comité específico dentro de la Agencia encargado de estudiar y supervisar esta información. También se revisa por el Comité de Medicamentos para uso humano el denominado SmPC —summary of products characteristics 52— o Ficha Técnica (FT) por su denominación en español, que incluye detalles sobre empaquetado y denominación.
El good clinical practice 53 engloba la presentación de estudios científicos y especializados que avalen los resultados de la investigación realizada respecto del futuro medicamento. En esta fase se suscitan algunas cuestiones que deben señalarse: la EMA solicita informes científicos a centros de investigación de renombre internacional —por ejemplo, el Instituto Karolinska en Suecia 54, el Centre Nationale de la Recherche Cientifique 55 en Francia, el Max Plank Institut 56 y la Helmholtz Gemeinschaft 57 en Alemania, o importante centro que depende de la Oxford University 58 en el Reino Unido—, respecto de los que se pueden suscitar siempre cuestiones de conflicto de interés, ya sea por su participación en programas paralelos de investigación, o por su vinculación con determinados programas nacionales, o con empresas o conglomerados farmacéuticos internacionales que participan en la subvención y financiación de medicamentos que, eventualmente, podrían entrar en competencia con aquellos respecto de los que se les está solicitando informe. Esta cuestión será suscitada en el siguiente capítulo.
De este modo llegamos a un hito en el camino del medicamento en la EMA que es la intervención del peer review 59, que se lleva a cabo en el seno del Comité de medicamentos para uso humano —CHMP—; esta revisión realizada por expertos se plasma en el nombramiento de un ponente y un co-ponente dentro del Comité (rapporteur y corapporteur), que deberán ser de países diferentes, y que llevarán a cabo la evaluación científica del medicamento de manera separada e independiente, para ello forman dos equipos de trabajos con expertos provenientes de las agencias nacionales europeas. Ambos grupos emiten dos informes exhaustivos que hacen referencia a una pluralidad de cuestiones relativas a la viabilidad del medicamento. Al mismo tiempo el CHMP nombra peer reviewers —revisores— que supervisen la presentación, argumentación, resultados y conclusiones de los informes de los grupos de trabajo coordinados por los ponentes.
Los resultados se valoran en una sesión plenaria del Comité de Medicamentos para Uso Humano, en la cual se suscitarán cuestiones relacionadas con la decisión definitiva a adoptar. Es importante indicar que con carácter habitual la EMA sigue el criterio expresado en sus informes tanto por el rapporteur principal como por el co-rapporteur, y esto especialmente es así en aquellos casos en los que ambos coinciden en sus conclusiones —ambos grupos trabajan de manera independiente y, por lo tanto, pueden llegar a conclusiones diversas—; por esta razón, cuando la Agencia se apartar de estas conclusiones necesitará una particular argumentación para avalar su decisión discordante, que avale el cambio de criterio. Recordemos, que en esa reunión se puede todavía solicitar asesoría científica complementaria de expertos ad hoc.
De este modo, se alcanza la resolución final del CHMP, que se lleva a cabo por consenso o unanimidad de forma habitual, siendo ésta la regla habitual, aunque hay un porcentaje (alrededor de un diez por ciento de los casos) en los que bastará con obtener la mayoría en el citado comité.
La EMA publica las agendas y minutas de las reuniones celebradas en los distintos comités una vez concluido el procedimiento 60, una vez recaída la resolución final sobre la solicitud de autorización de comercialización. Este documento se denomina European Public Assessment Report (EPAR) 61.
Respecto al calendario de aprobación de una solicitud de comercialización de un medicamento ante la EMA, la Agencia declara que requiere hasta 210 días “activos”, plazo que se puede ver interrumpido por dos clock-stops 62, que son dos plazos que se conceden al solicitante para que responda a las preguntas que se le hayan planteado en el cuestionario del CHMP, y que puede durar hasta seis meses, y un segundo plazo de hasta tres meses en el que debe dar respuesta a la lista de cuestiones pendientes —list of outstanding issues—. En total se identifican 21 pasos distintos dentro del complejo calendario del procedimiento ante la Agencia, que —en la mayoría de los casos— hace que el conjunto temporal del proceso de evaluación pueda superar los dos años de duración antes de que se emita la opinión o decisión final 63.
2.4. Autorización
Conforme a la legislación vigente 64 y al estatuto jurídico de la EMA, la resolución final vinculante respecto a una solicitud de autorización de comercialización de un medicamento corresponde llevarla a cabo a la Comisión Europea, a propuesta de la Agencia, que se limita a emitir una recomendación para que la Institución de la que depende orgánicamente dicte la decisión definitiva en el plazo de 67 días. Estas resoluciones se publican y constan en un sitio internet de la Comisión Europea 65.
Ante la resolución, en la medida en que se trata de un acto definitivo, se podría eventualmente suscitar un recurso de apelación ante el
Tribunal General de Justicia de la Unión Europea, ya que es la jurisdicción pertinente para conocer de los pleitos que se planteen contra actos y decisiones definitivas emanadas de la Comisión Europea.
2.5. Acceso
Una vez concedida la autorización se suscita la cuestión de su posicionamiento comercial, en concreto, del precio: en este ámbito la EMA sólo ofrece cierto apoyo a los órganos de asesoramiento tecnológico —Health Technology Assessment bodies (HTA)—, que trabajan en colaboración con las agencias nacionales correspondientes; el titular de la autorización puede decidir en qué países de la UE va a ofertar el medicamento ya que no está obligado a comercializarlo en todos los Estados miembros, y también determinar que países en el ámbito internacional son los más atractivos para su implantación.
2.6. Monitorización de la seguridad
El último paso que realiza la EMA consiste en la evaluación del medicamente respecto a su seguridad, tarea que se despliega en los países en los que se ha comercializado. La Agencia cuenta con un órgano especializado en esta labor —el Comité de valoración de riesgo en farmacovigilancia (PRAC) 66-, aunque esta supervisión puede realizarse a solicitud de un Estado miembro o de la propia Comisión Europea. El fin es llevar a cabo una mejor coordinación a nivel europeo de las consecuencias de un determinado fármaco o medicamento.
3. ASPECTOS PROBLEMÁTICOS: TRANSPARENCIA, CÓDIGO DE CONDUCTA Y CONFLICTOS DE INTERÉS
La transparencia es un concepto abierto, jurídicamente indeterminado, con caracteres de difícil pormenorización y explicitación, con connotaciones plurales, pero siempre de importancia básica radical, porque explicita algo que se ha convertido en la manifestación de una forma de pensamiento, de una manera de actuar, de entender la función y el servicio públicos, y, por lo tanto, de superar la consideración del ciudadano como administrado, como sujeto pasivo de procedimientos administrativos, de ente meramente sometido al Derecho público, donde se parte siempre de la concesión de prerrogativas para la administración correspondiente, con independencia que sea nacional o forme parte de las Instituciones Europeas y de sus organismos y agencias dependientes —como es el caso presente—. Además, esas facultades exorbitantes propias de la Administración, que se manifiestan —entre otras potestades— en la ejecución y autotutela de los actos administrativos o en la presunción de veracidad y de conformidad a Derecho de los procedimientos incoados hacen que difícilmente el sujeto pasivo de los mismos, el ciudadano, pueda gozar de una situación de igualdad en relación con el órgano administrativo que incoa o instruye un procedimiento, con independencia de la naturaleza del mismo.
Por eso, la apelación a la transparencia no puede ser un concepto vacío, un expediente trivial y superficial, que simplemente reconozca algunas potestades al administrado respecto de la acción de las administraciones públicas: la transparencia implica un cambio de paradigma, de mentalidad, de forma de hacer las cosas, y debe alcanzar e impregnar todos los entes, organismos y agentes que manifiestan, de una forma u otra, potestades públicas; de aquí que reducirla a la publicación de determinada información y de determinados documentos evidenciaría una visión escueta, rala y reduccionista de la transparencia.
La Unión Europea lleva embarcada muchos años en la implementación progresiva de políticas de transparencia, en este sentido se puede citar como hito relevante el Libro Blanco sobre la Gobernanza Europea 67, publicado en 2001, y que se enmarcó dentro de la ambiciosa Estrategia de Lisboa del año 2000, y que buscaba —entre otras cuestiones— acercar las Instituciones Europeas y sus procedimientos a los ciudadanos, como auténticos protagonistas de la integración europea, así se expresaba el Libro Blanco afirmando que
El Libro Blanco propone abrir el proceso de elaboración de las políticas de la Unión Europea con el fin de asociar a un mayor número de personas y organizaciones en su formulación y aplicación, lo que se traducirá en una mayor transparencia y en una mayor responsabilización de todos los participantes. Esto debería permitir a los ciudadanos comprobar cómo los Estados miembros, actuando de manera conjunta en el marco de la Unión, son capaces de responder más eficazmente a sus preocupaciones.
De este modo, progresiva y paulatinamente la transparencia se ha convertido en una auténtica política europea, que se promueve especialmente desde la Comisión Europea 68, y que se fomenta a través de diversos capítulos o líneas de actuación, como son los Códigos de Conducta —para los propios miembros y agentes de la Comisión— y el Código de Buena Conducta Administrativa —al que haremos referencia en lo sucesivo—, la creación de un Registro de Transparencia para los interlocutores sociales y económicos que quieren interactuar en los procesos comunitarios, o también la publicación del Acuerdo Institucional que implica tanto al Consejo, al Parlamento Europeo y a la propia Comisión Europea en la implementación de la transparencia 69.
La Agencia Europea de Medicamentos, como hemos indicado, es una agencia descentralizada, con potestades ejecutivas y reglamentarias, de manera que —como bien se indica en su estatuto fundacional— forma parte del entramado institucional de la Unión Europea, ostentando personalidad jurídica autónoma, y, por lo tanto, teniendo que acomodar sus procedimientos, actuaciones y régimen propios a la regulación vigente en materia de transparencia para todos los órganos comunitarios. Además, explícitamente se equipara a sus funcionarios, personal y agentes con los de las Instituciones Europeas, sometiéndoles al mismo estatuto y régimen jurídico 70.
Se puede afirmar que, desde esos momentos iniciáticos de los años 2000 en relación con este principio se ha recorrido mucho camino, y es conveniente reseñar que algunas de las cuestiones que entonces se suscitaron ya se han incorporado a las prácticas administrativas de las Instituciones Europeas, y —por lo tanto— el camino emprendido se puede calificar de alentador, aunque —como vamos a ir poniendo de manifiesto—, es necesario que esta tendencia se propague en todas las agencias descentralizadas y órganos consultivos; ya que, de otro modo, y dada la progresiva y paulatina tendencia a la traslación expansiva de potestades y competencias respecto de estos órganos, podrían quedar desvirtuados algunos de los elementos atinentes a la transparencia que ya están siendo implementados en las Instituciones de la UE.
A continuación, vamos a proceder a un análisis en cierta profundidad de aquellos puntos y elementos que se suscitan respecto a la transparencia en la Agencia Europea de Medicamentos, para así alcanzar una perspectiva que permita también comparar los métodos de la EMA con parámetros vigentes en otras organizaciones asimilables en otras jurisdicciones, como puede ser concretamente la Food and Drug Administration en los Estados Unidos, siempre con el objetivo constructivo de formular propuestas de mejora respecto de la situación actual.
En primer término, y a efectos de exposición de la cuestión, conviene distinguir entre los elementos que son más relevantes a efectos de la consecución de un ambiente proclive a la transparencia en una organización, y —por supuesto— también en la Agencia Europea de Medicamentos: de este modo, tendremos que contemplar las prácticas relativas a la publicidad y al acceso de documentos de los comités, grupos de trabajo y órganos de gobierno que la integran. En segundo lugar, nos fijaremos en el estudio de los Códigos de conducta vigentes en el ámbito de actuación de la EMA. Por último, nos centraremos en los conflictos de interés, y todas las cuestiones atinentes a su detección temprana, gestión y evaluación.
3.1. Publicidad de la información y acceso de documentos
En primer lugar, hay que indicar que la base normativa fundacional de la EMA —el Reglamento 726/2004— recoge la transparencia como principio que debe guiar las actividades de la Agencia: en concreto, se establece que el Consejo de Administración, a propuesta del Directo Ejecutivo y en coordinación con la Comisión, adoptará normas para la puesta a disposición del público de informaciones reglamentarias, científicas o técnicas, relativas a la autorización y al control de medicamentos, que no tengan carácter confidencial 71, además se obliga a que el reglamento interno y los procedimientos de la EMA sean asequibles y accesibles al público a través de su sitio internet.
Por otra parte, existe un documento interno de la Agencia que recoge los principios que deben regir la publicación de las agendas y de las minutas y actas de los comités científicos 72. Comienza este documento alegando la base jurídica de la obligación de publicidad, haciendo referencia a los artículos 26 y 80 del Reglamento 726/2004. A su vez, el artículo 66 de éste hace referencia a la obligación del Consejo de Administración de aprobar normas para garantizar que la información relativa a la autorización o control de medicamentos se hace pública.
En relación con la publicidad de información a nivel del conjunto de las Instituciones Europeas y de todos los organismos dependientes, la norma de referencia es el Reglamento 1049/2001, relativo al acceso público a los documentos de los mismos 73. Esta norma parte de la afirmación del principio de apertura que permita a los ciudadanos una mayor participación, aportando así legitimación, democracia y legitimidad a la acción de las Instituciones Europeas 74. De este modo la EMA ha llevado a cabo una serie de acciones en relación con el cumplimiento de los objetivos que se fijaban en esta disposición.
Como hemos indicado en primer término, en 2013 se publicaron los Principios de publicidad de los actos relativos a los Comités científicos de la Agencia, que se pueden resumir en los siguientes:
— Transversalidad de la obligación de transparencia respecto de todos los comités científicos.
— El punto de partida —que debe servir de criterio en relación con la publicidad de las agendas— es que todo su contenido debe ser accesible, aunque con las limitaciones derivadas de la confidencialidad comercial y de la protección de datos de carácter personal; también se introducen otros criterios restrictivos derivados del mantenimiento del orden público, para no suscitar situaciones de alarma social. Sin embargo, nos encontramos ya con un primer elemento que suscita ciertas dudas, al afirmarse que la publicación de la información debe llevarse por medio de la menor redacción posible 75. Se nos antoja interesante esta matización, en la medida en que parece poco coherente exigir un alto nivel de transparencia, recurriendo a la técnica de la mínima manifestación de la misma.
— Se excluye de la publicidad la información relativa a las consultas científicas y a las solicitudes de protocolos de asistencia —Protocol Assistance requests—.
— Se entiende que la salvaguarda de la información calificada de comercialmente confidencial —commercially confidential— es un bien susceptible de respeto y protección, de manera que se establecen trabas relevantes respecto a su comunicación pública.
— Los nombres de los ponentes y co-ponentes deben hacerse públicos. Esta obligación puede parecer evidente, pero es de naturaleza sustancial, al basarse el Comité de Medicamentos para Uso Humano en sus conclusiones para elaborar su propuesta de resolución.
— El principio que quiere transmitir la EMA es que su nivel de transparencia y de calidad de acceso a los documentos relativos a sus procedimientos es superior que la que impera en el resto de las Instituciones y organismos comunitarios.
— Se entiende que la publicidad de las minutas y actas de reuniones —tanto del Consejo de Administración, como de los Comités científicos y grupos de trabajo— debe limitarse a ofrecer una buena calidad de comunicación que refleje los resultados alcanzados en las mismas. 76 Queda por determinar el significado de ese matiz que exprese una buena calidad de la información, ya que una interpretación restrictiva del concepto podría dejar sin sentido la obligación de transparencia.
— Las opiniones y reflexiones personales emitidas durante las reuniones anteriormente expresadas no tienen por qué recogerse, ni ser publicadas. Sin embargo, si deben publicarse de manera completa la lista de participantes, los intereses que hayan declarado, y las restricciones a que estén sujetos.
A su vez los Principios de publicidad de la EMA conforme al Reglamento 1049/2001 hacen referencia a la comunicación de determinados hitos del procedimiento de evaluación: de esta forma, respecto de las opiniones previas en las solicitudes de autorización de comercialización —Pre-opinion in the framework of marketing authorisation applications—, el principio que rige es el de que las minutas sólo reflejen el resultado de los items más importantes tratados. Respecto de la opinión en el ámbito de las mismas —Opinion in the framework of marketing authorisation applications— se deberá publicar el resultado de las votaciones que hayan tenido lugar y también los nombres de los que votaron de manera divergente, salvo que se solicitase un proceso de nuevo examen, en cuyo caso los que hayan votado contrariamente no se harán públicos hasta que conste el resultado de ese nuevo proceso.
Por su parte, en 2006, el Consejo de Administración de la EMA aprobó las denominadas Normas 77 para la implementación del Reglamento 1049/2001 78. Este documento contiene elementos que suscitan nuestro interés en relación con la comunicación y publicidad de ciertos documentos de la Agencia, ya que recoge explícitamente las excepciones a la publicidad de algunos de estos, de modo que en Anexo se enumeran las normas que permitirán declarar determinada documentación como secreta o clasificada.
1) En concreto, la Agencia podrá denegar el acceso a un documento cuando pueda minar la protección del interés público en relación con la seguridad, la defensa militar, las relaciones internacionales, la política financiera, monetaria o económica de la UE o de un Estado miembro; también se puede denegar cuando pueda afectar a la integridad y privacidad de un particular, en especial en relación con los datos de carácter personal protegidos conforme a la legislación vigente en la UE 79.
2) También se podrá denegar el acceso en el supuesto de que su publicidad pueda afectar a la protección de los intereses comerciales de personas físicas o jurídicas, incluyendo la propiedad intelectual; o cuando hagan referencia a cuestiones que estén siendo objeto de un proceso judicial, o siendo sometidas a inspección, investigación o auditoría. Aunque esta limitación se somete a la cautela de que pueda prevalecer un interés público superior que exija su divulgación 80.
3) Si la documentación relevante hace referencia a un procedimiento que esté pendiente ante la EMA, podrá denegarse su acceso y comunicación, en la medida en que la misma pudiese afectar gravemente al proceso decisorio 81.
4) Se recoge también una excepción de comunicación que —en nuestra opinión— es bastante relevante: se podrá denegar el acceso a un documento que contenga opiniones emitidas en las deliberaciones y en las consultas preliminares en el seno de la Agencia, de manera que no podrán ser conocidos ni siquiera con posterioridad a la toma de la decisión correspondiente, cuando se entienda que podría minar los procedimientos internos de la misma 82.
5) También se podrá denegar la comunicación de documentos que procedan o sean propiedad de terceros 83.
6) En el caso de que el documento provenga de un Estado miembro, éste deberá ser consultado con carácter previo a su divulgación 84.
7) En el supuesto de que un documento sólo con carácter parcial estuviese sometido a alguna de las excepciones expuestas con anterioridad, podrá publicarse aquella parte del mismo que no quedase afectada por la restricción pertinente 85.
Por su parte, el Anexo de las Normas que estamos considerando recoge aquellos documentos que pueden ser calificados como confidenciales y restringidos:
— Documentos restringidos serían aquellos cuya publicación no autorizada podría afectar de manera perjudicial a los intereses de las Instituciones Europeas, de los Estados miembros y de la propia Agencia; su característica fundamental reside en que eventualmente podrán ser puestos en conocimiento del público, pero su publicidad prematura podría implicar perjuicios a los intereses de los actores expresados con anterioridad, así como a los solicitantes y organismos implicados en la concesión de autorizaciones de comercialización. De esta manera, con carácter general, el concepto de documentos restringidos se restringirá a los supuestos expuestos con anterioridad en las seis categorías recogidas en el artículo 3 del Reglamento.
— Por su parte, se declarará confidencial aquella información que pudiese perjudicar los intereses esenciales de las Instituciones Europeas, de los Estados miembros o de la Agencia; estos necesariamente deben poder englobarse en alguno de los apartados del precepto citado con anterioridad 86.
La consideración de todas y cada una de las excepciones a la publicidad o comunicación de documentos de la Agencia Europea de Medicamentos implicaría un exhaustivo trabajo de remisión a circunstancias y hechos particularizados, que excederían estas páginas, pero sí es necesario manifestar y concretar algunas dudas relevantes que prima facie se nos suscitan en términos generales. Por un lado, entendemos que las excepciones recogidas en el artículo 3 son muy amplias, se basan en el recurso a conceptos jurídicos indeterminados que son de difícil precisión y más compleja concreción: la apelación al interés público que se extiende hasta la protección del mismo en el marco de la política financiera, económica y monetaria nos parece algo exorbitante, porque engloba parámetros de restricción de la comunicación que exceden indudablemente la labor central y objetiva de la Agencia, que reside en la autorización de comercialización de medicamentos.
Es evidente que el gasto sanitario derivado de la aprobación de comercialización de algunos medicamentos puede tener consecuencias sobre los presupuestos de los Estados miembros, pero —al mismo tiempo— la ponderación del bien jurídico con el que nos vemos contrastados también hace referencia al bien común, e incluso puede ser considerado superior a los postulados derivados de la mera política económica y presupuestaria, ya que ese mismo bien común exige la salvaguarda de la salud, de la salus publica, que incluso puede llegar a considerarse como presupuesto del interés general, como tan crasamente ha puesto de manifiesto la pandemia reciente de la COVID 19.
También nos parece que la excepción que deniega la comunicación de deliberaciones relativas y que se suscitan en el ámbito de la toma de decisiones dentro de un debate relativo a la autorización de comercialización de un medicamento, es algo incoherente con el concepto mismo de transparencia, porque justamente lo que se busca y persigue con la publicidad de estas consultas y debates —incluso preliminares— es aportar claridad a los criterios que han contribuido a la formación de la decisión de la Agencia en cuestión tan trascendente —no olvidemos que sus decisiones determinan la autorización por el resto de las Agencias nacionales de un determinado medicamento, y también sirve de referencia respecto de su comercialización a nivel internacional—. De esta forma, la facultad de restringir el acceso al contenido de estos debates, por un lado, oscurece la transparencia del procedimiento, y —por otro lado— no contribuye en absoluto a la implementación de una cultura de transparencia que supuestamente se declara transversal para todas las actividades de la EMA —como ostentosamente se expresa en el primer capítulo relativo a los objetivos de la Agencia 87—.
Por su parte, la distinción que se lleva a cabo en el Anexo relativo a los documentos que pueden ser declarados restringidos o confidenciales, basándose única y exclusivamente en el criterio de la afectación a los intereses de la Agencia, de las Instituciones Europeas o de los Estados miembros y su calificación de esencial, nos parece realmente sorprendente. La primera cuestión que se nos suscita es la relativa a la ponderación de esa esencialidad de la afectación que indudablemente recae en la propia Agencia —en concreto, en el Consejo de Administración—. Este órgano está compuesto por representantes nombrados por los Estados miembros, de manera que las implicaciones de la declaración del carácter esencial siempre va a tener connotaciones o consideraciones de carácter político en su manifestación, siendo que la calificación de un documento como confidencial o restringido es diferente, ya que esta información puede llegar al público y a los interesados en algún momento —superadas las apreciaciones que provocaron su calificación de restringidos—; sin embargo, la declaración de confidencialidad por afectación del interés general excluye de comunicación y de acceso, y por lo tanto, deniega con carácter permanente su accesibilidad.
También se nos antoja exorbitante la excepción de comunicación cuando la información proceda de un tercer interesado que ha participado en un procedimiento ante la EMA; en la medida en que se supedita a la consulta a esa parte interviniente, se lleva a cabo una atribución tácita a un tercero de poder disponer sobre lo que constituye un auténtico derecho que corresponde a todos el público en general en la medida en que participen de la ciudadanía europea: el derecho a la transparencia.
No parece superfluo recordar, en este sentido, que el derecho de acceso a los documentos se configura como derecho básico de los ciudadanos europeos en la Carta de los Derechos Fundamentales de la UE 88, este derecho se enmarca en el ámbito de los relativos a una buena administración. No es una declaración baladí, fútil o vacía de contenido, ya que otorga potestades subjetivas que acrecen el patrimonio jurídico del ciudadano singularizado, y cuya limitación debe someterse a criterios reconocidos tanto en la jurisprudencia europea como en la emanada por el Tribunal de Derechos Humanos de Estrasburgo, de forma que la afectación del derecho no puede suponer en ningún caso su crasa negación, ni la afectación del contenido esencial que le hace reconocible, porque de otro modo la transparencia se convertiría en una mera declaración insustancial, vacua, y sin trascendencia jurídica alguna.
2.2. Códigos de conducta
Antes de nada, decir que respecto de esta Agencia europea descentralizada concurre un hecho singular, por único en el panorama comunitario: la EMA está sometida a tres Códigos de conducta que conviven y tienen pretensión de vigencia en el marco de sus actividades: en primer lugar, la Agencia está sometida al Reglamento interno 89 de la Comisión Europea —al depender orgánica y jerárquicamente de esta Institución—, en el que se explicitan las obligaciones del personal al servicio de la misma, y también las limitaciones que tienen respecto a una serie de elementos —actividades, conflictos de interés, regalos, ocupaciones post cargo, transparencia, etc.—, en esta disposición nos encontramos con un anexo en el cual se recoge un documento denominado Código de Buena Conducta Administrativa para el personal de la Comisión Europea en sus relaciones con el público —que conforme a lo prescrito en el artículo 75 del Reglamento 726/2004 y en el propio Código de Conducta 90, es aplicable plenamente al personal de las agencias y organismos dependientes de la Comisión al equipararlo con los funcionarios dependientes de aquella—, en este Código se plasma una serie de obligaciones relativas a la relación que deben tener los agentes y funcionarios con los particulares, de manera que en el capítulo dedicado a la información de las partes interesadas se declaran las siguientes obligaciones 91: conceder audiencia de todas las partes con interés directo, la obligación de motivar las decisiones y resoluciones, y también la de indicar los posibles recursos.
Este Código dedica una parte relevante de su articulado a la regulación de la seguridad en la Comisión Europea, incluyendo criterios de determinación de la oportunidad de calificar un documento como clasificado 92, indicando que se requieren cierta cautela y experiencia a la hora de seleccionar la información y el material que vayan a protegerse.
Por su parte, el Código de Buena Conducta administrativa de la EMA 93 supone el reflejo a nivel de ésta de los principios expresados en el más genérico de la Comisión, adaptándolos a las necesidades propias de la misma, e incluyendo algunas cuestiones que merece la pena ser reseñadas.
En primer lugar, extiende el Código su radio de acción a una amplia categoría de sujetos, en la medida en que entiende que quedan vinculados por el mismo no sólo los agentes y funcionarios al servicio de la EMA, sino también los expertos nacionales, el personal en prácticas, aquellos que tengan contratos en interinidad con contratos de régimen laboral, los expertos visitantes y los demás asistentes científicos con independencia de su vinculación, y a los que en su conjunto califica de staff 94.
Hace especial énfasis este Código en la imparcialidad e independencia del personal de la EMA, de manera que incluye un interesante baremo para su determinación: el respeto a los principios de la integridad científica 95, de manera que deberán abstenerse de manifestar o incurrir en cualquier acto que pudiese denotar un trato preferente o de favor, en especial en relación con los intereses nacionales y políticos de los países de los que fuesen nacionales, o cuestiones de naturaleza personal 96.
También nos parece oportuno citar el apartado 8 de este Código de conducta administrativa, que hace referencia al importante punto de la coherencia y las expectativas legítimas 97 suscitadas en los que se someten a los procedimientos de autorización de la Agencia; la confianza legítima es un principio de derecho administrativo, según el cual el administrado puede confiar en que la Administración actuará de manera conforme a como ha venido resolviendo en casos o situaciones asimilables o parecidas. De este modo, se crea esa expectativa que obliga a la Administración —en este caso a la EMA— a seguir la praxis o línea de actuación que haya sido habitual en circunstancias equiparables. De hecho, en el supuesto de que vaya modificar esa costumbre, deberá hacerlo sólo ante hechos que aconsejen un cambio o modificación en su criterio: además, esto tiene que estar justificado, y responder a una fundamentación y razonabilidad que aporten convicción al cambio de criterio.
Por ejemplo, esta circunstancia podría suscitarse en el supuesto de que la Agencia no siguiese, en la resolución de una solicitud de autorización de comercialización, el dictamen de los ponentes y co-ponentes, especialmente cuando en circunstancias previas equiparables se hubiese constatado que habitualmente se había aceptado la línea asumida en la ponencia por el rapporteur y co-rapporteur.
Es importante señalar estos matices, ya que —en caso contrario— la exposición de los parámetros de buena administración se verían limitados y restringidos a meros principios formalistas, que no afectarían al fondo de las cuestiones ventiladas, y, por lo tanto —como hemos indicado— no podrían hacer surgir auténticos derechos subjetivos en los administrados. En este sentido se enmarca también el principio de proporcionalidad 98 que se declara en el apartado 4 del Código, y que exige una proporcionalidad entre los límites impuestos a los solicitantes, y el interés público subyacente, para encontrar un correcto equilibrio entre las decisiones adoptadas y la restricción impuesta sobre las expectativas de los particulares partícipes en el procedimiento.
De nuevo, la relación entre ambos principios, el de proporcionalidad, y el de respeto de las expectativas legítimas, siempre es muy estrecha, ya que hacen ambos referencia al alcance y pertinencia de las medidas que pueda la Agencia tomar en relación con los objetivos perseguidos, es decir, entre el interés público que se manifiesta en la seguridad ante la aprobación de un medicamento, y las perspectivas suscitadas en el particular.
Conviene citar otro documento relevante a los efectos que estamos considerando: el Código de Conducta de la Agencia Europea de Medicamentos 99,que se aprobó siguiendo el imperativo expresado en el Reglamento de referencia de la misma 100, y que obliga a que los miembros de la Agencia se sometan a una sería de cautelas para evitar que se puedan suscitar conflictos de interés, a través de la imposición de la obligatoriedad respecto del personal y agentes de la EMA de presentar declaraciones anuales en las que manifiesten voluntariamente los eventuales conflictos en que hayan podido incurrir.
De este modo, el Código de Conducta se hace imperativo para todo su personal —staff—, y también obliga a los asesores externos que puedan participar en sus procedimientos. Se busca alcanzar un estándar elevando en este ámbito, y, por lo tanto, se exige la mayor transparencia posible.
Este Código se articula alrededor de cuatro apartados, tres de los cuales están dedicados a los conflictos de interés, al deber de confidencialidad y de discreción, y el cuarto a los criterios relativos a regalos e invitaciones. Es cierto que el Reglamento 726/2004 hace una referencia explícita a los conflictos que se pudiesen derivar de intereses relacionados con la industria farmacéutica, mientras que el Código exige también que se actúe con lealtad e integridad.
En este sentido, la EMA lleva a cabo un esfuerzo importante en la determinación del concepto de interés, y también de la de la noción de interés directo: en cualquier caso —ya sea por explícita relación o conexión con algo que puede prima facie suscitar un conflicto de interés, o simplemente por ostentar una relación, cualidad o circunstancia que pueda influir sobre la labor del personal afectado— la realidad es que la Agencia somete a ponderación cada caso de forma singularizada, para interpretar las limitaciones que haya de imponerse a la actuación del sujeto afectado, pudiendo llegar a impedirle votar o participar en reuniones relevantes.
En este sentido, se pueden distinguir tres tipos de declaraciones: la declaración inicial que tienen que llevar a cabo todos los empleados, trabajadores y expertos que colaboren con la EMA; la declaración espontánea —que se suscita voluntariamente cuando se perciba que se puede incurrir en un eventual conflicto de interés con carácter sobrevenido—, y las declaraciones de actualización que tienen carácter anual como mínimo.
En las Staff Regulations 101 se recogen las consecuencias que se pueden derivar de los incumplimientos relativos a las declaraciones de intereses, de manera que pueden acarrear incluso la prohibición de trabajar para la Agencia; además, las declaraciones individuales deben ser asequibles para su consulta en el sitio internet de la Agencia, para que las partes interesadas puedan ser conscientes de eventuales circunstancias que fuesen relevantes respecto de sus solicitudes.
En este sentido, quisiéramos matizar una cuestión: por un lado, la veracidad de una declaración depende esencialmente de la voluntad del sujeto afectado por el conflicto de interés, que, en algunas ocasiones, puede suscitarse con carácter sobrevenido ante nuevas circunstancias; de este modo, se parte de la presunción de buena fe, algo que puede resultar arriesgado, y ciertamente de compleja prueba y alegación. Por ejemplo, la cercanía a institutos de investigación, a centros dependientes de Universidades, a grupos de trabajo nacionales o de las propias agencias nacionales, así como los que podríamos calificar de inputs imponderables, como pueden ser la alineación de países con posturas y mentalidades con cierta similitud respecto de solicitantes que no procedan de su ámbito de influencia, los prejuicios respecto de determinadas concepciones científicas; en fin, los elementos de ponderación se nos antojan tan plurales, y hasta cierto punto inasequibles, que suscitará alguna consideración en el capítulo relativo a las propuestas de mejora de la Agencia.
En cualquier caso, no deja de sorprender que el Código no recoja la abstención del afectado por un conflicto de interés como primera obligación, de manera que se requiere la intervención de la Agencia para calibrar el alcance del conflicto de interés, y las consecuencias que haya que sacar del mismo a efectos del procedimiento en cuestión.
En cualquier caso, nos parece oportuno reseñar el enfoque —en nuestra opinión, oportuno— que se lleva a cabo en las Reglas de Procedimiento respecto del Comité para Medicamentos de uso humano, que parte de una idea que nos parece elogiable, al incidir en las garantías de independencia, de manera que se combina la publicidad de las declaraciones con la que se lleva a cabo de los currículos y trayectorias profesionales, algo que —en muchas ocasiones— puede servir para poder discernir si alguien puede estar incurso en un conflicto de interés; además, se obliga a que antes de cada reunión del comité se declaren posibles líneas de incompatibilidad por parte de los asistentes respecto del asunto a tratar en el orden del día. 102
El otro gran apartado en el Código de Conducta es el relativo a la Confidencialidad y a la Discreción, que en parte está relacionado con el acceso a los documentos por parte del staff de la Agencia y por los particulares interesados, y también a la clasificación de aquellos —algo a lo que ya nos hemos referido con anterioridad—. La EMA tiene un protocolo para dar respuesta a las solicitudes de acceso a información 103, que esencialmente se basa en un procedimiento escrito, en el que se deben exponer con veracidad y claridad los intereses representados, y los procedimientos afectados.
Es importante señalar que siempre se parte de la obligación de confidencialidad, algo que ya se exigía en el Reglamento 726/2004 104 respecto de los empleados de la EMA, y además está en coherencia con las Staff Regulations aplicables a todos los funcionarios y empleados de las Instituciones Europeas 105, aunque —correctamente, en nuestra opinión— también se reconoce un derecho a la libertad de expresión de los mismos, en la medida en que no provoque situaciones de conflicto o rupturas inasumibles de la confidencialidad, o cuando se puedan provocar daños respecto de intereses de terceros, en procedimientos pendientes de resolución, o para los Estados miembros o las propias Instituciones Europeas 106.
Por otra parte, es innegable que la información que se maneja por los funcionarios y empleados de la EMA en muchas ocasiones, por su propia naturaleza, es confidencial: información comercial, datos relativos a líneas de investigación, compromisos internacionales, posiciones de los Estados miembros, de manera que es difícil alcanzar un equilibrio entre la transparencia, la libertad de comunicación, y la salvaguarda del interés general comunitario, manifestado en la acción de la Agencia en relación con los procesos de autorización de comercialización de medicamentos, que es algo de una trascendencia fundamental para toda la UE.
2.3. Gestión de conflictos de interés en la EMA
Es evidente que, entre todos los organismos públicos que quedan incardinados en el marco de las Instituciones Europeas —ya sean agencias ejecutivas, descentralizadas, comités consultivos u órganos independientes— la EMA quizá sea en la que una mayor cantidad de conflictos de interés se puedan suscitar. Esto se deriva de la propia naturaleza de sus funciones, y de la confusión que se produce entre procedimiento de autorización y asistencia técnica, que se origina y se ofrece desde la misma Agencia.
Este punto es de absoluta relevancia en nuestra opinión: la Agencia debe cohonestar dos funciones que son radicalmente complejas, y que —en línea de principio— no tienen por qué ser compatibles: emitir un juicio de comercialización, y al mismo tiempo, seleccionar quién lleva a cabo el expertise y la asistencia técnica para poder desarrollar esta función de estudio y análisis, son dos actuaciones distintas, complementarias, pero que si se llevan a cabo por la misma y única entidad —la EMA— puede suscitar dudas y cierta lógica prevención.
Esencialmente deberían ser dos funciones claramente diferenciadas, sometidas a exigencias y procesos distintos, y que sólo en su fase final deben de coincidir. La apelación a una pluralidad de expertos en especialidades médicas diversas, vinculados necesariamente a centros de investigación, empresas, universidades, que pueden trabajar para una pluralidad de intereses, y que —per se— tienen perspectivas distintas en relación con las políticas de transparencia y de conflictos de interés, sólo puede suscitar y conllevar situaciones complejas en relación con la labor de la EMA, siendo que el mero recurso a la declaración anual de intereses queda reducido a algo cuasi anecdótico ante la complejidad de circunstancias subyacentes que pueden influir en la actividad de ese experto, ya sea externo o partícipe de la estructura de la EMA. Además, esta no deja de ser una técnica ex ante, es decir, la persona que lleva a cabo la declaración no puede prever, en el momento que la lleva a cabo, todas las circunstancias que, de forma sobrevenida, y respecto de un concreto y singular procedimiento, se puedan eventualmente suscitar —incluso atendiendo a meras tesituras personales—.
Si a esto le sumamos la vinculación que se produce con los Estados que proponen a estas personas —recordemos que esto es evidente y explícito tanto respecto de los miembros del Consejo de Administración como de los componentes del Comité de Medicamentos de uso humano—, la realidad es que la complejidad y multiplicidad de situaciones es casi inabarcable, y también resulta evidente la tremenda dificultad de poder afrontar criterios y baremos de carácter general, que pudiesen englobar la mayor cantidad posible de escenarios imaginables.
Por esta razón, la EMA de forma congruente con el enfoque asumido del recurso a una mera regulación formal de las situaciones a tratar, también mantiene este criterio respecto de los conflictos de interés. Así nos encontramos con una serie de disposiciones internas de la Agencia que inciden eminentemente sobre elementos objetivos, y que intentan ofrecer una respuesta a estas complejas circunstancias. Pero no podemos perder de vista que del tratamiento que se dé a esta problemática, depende la propia efectividad, eficacia, e incluso credibilidad de la Agencia en su conjunto.
En relación con la materia que estamos tratando, nos encontramos con dos importantes documentos que conviene reseñar por su trascendencia y alcance: la European Medicines Agency Policy on the handling of competing interests of scientific committees’ members and experts 107 —también conocida como Policy 0044—, y la Guía de procedimiento respecto a la inclusión de intereses declarados en el formulario electrónico de declaración de intereses para los miembros de comités científicos y expertos 108. La Policy 0044 entró en vigor el 1 de enero de 2021, y sustituye varias normas que habían estado vigentes desde 1995 en la Agencia respecto de esta materia.
Esta Policy 0044 parte de ofrecer unas definiciones de carácter general respecto a qué haya que entender bajo conceptos tan relevantes como son el sector biotecnológico, empresa farmacéutica, o del sector de la medicina. También lleva a cabo una distinción entre intereses directos e indirectos, calificándolos en una u otra categoría dependiendo del nivel de vinculación con la empresa o sector que suscite el interés. En concreto, entiende que hay intereses directos cuando se esté en alguno de los siguientes supuestos:
— Empleo en una empresa farmacéutica, del sector de la biotecnología o de la medicina.
— Consultoría respecto de las categorías de empresas citadas con anterioridad.
— Asesor estratégico en este tipo de empresas
— Ostentar intereses financieros en estas compañías.
— Cuando exista una implicación del experto en el reposicionamiento de productos médicos 109.
Sin embargo, los intereses indirectos se categorizan en función del empleo desempañado: Investigador principal, investigador, investigador clínico, becario, implicación en la organización del experto, vinculación familiar estrecha.
Por otra parte, es interesante reseñar algo que se manifiesta en los objetivos declarados como prioritarios en esta reglamentación 110: hay que intentar alcanzar un equilibrio o término medio entre la necesidad de ofrecer una garantía de independencia respecto del experto que vaya a participar en la labor de los distintos comités de la agencia, y la de ofrecer una asistencia o expertise de calidad: así se usa la expresión cooling-off period para designar ese período de tiempo transcurrido el cual se podría entender que el conflicto de interés ya ha perdido relevancia, permitiendo así la implicación del experto en el procedimiento en cuestión.
De esta forma, y distinguiendo conforme al tipo de interés declarado, y el cargo desempeñado, se enumeran en la norma hasta diez tipos distintos de restricciones con sus correspondientes periodos de duración. No corresponde a este trabajo entrar en una ponderación de los escenarios que se recogen en la Policy 0044, pero sí nos parece obligado expresar una opinión respecto de la técnica utilizada, que se basa en la apelación a elementos formalistas, derivados tanto de las categorías y grados de implicación en los sectores afectados de los expertos, como de la duración de su implicación: nos causa cierta perplejidad, en la medida en que no nos parece que se cubran todas las circunstancias de las que se pueden derivar conflictos de interés —por ejemplo, se omite la afinidad nacional, la vinculación derivada de proyectos conjuntos, de programas participados, de financiación compartida, de situaciones derivadas del desarrollo de medicamentos competitivos etc.—, y —reiteramos— se omite cualquier referencia a la afinidad política o gubernamental, que quizá pueda ser relevante en algunos casos y respecto de algunas personas.
Por todo ello, seguimos sosteniendo que la pretensión de encuadrar en categorías cerradas prefijadas la multiplicidad y variedad de circunstancias es una pretensión que puede resultar baladí, vacua y fútil: por eso, enfatizar en las técnicas de inclusión en el modelo electrónico de declaración de intereses del staff de la Agencia y sus colaboradores, no hace más que abundar en esta idea de la primacía de lo formal sobre el elemento material, ya que la declaración de intereses tiene, por su propia naturaleza, un aspecto intangible, y de compleja aprehensión y determinación, al depender en gran parte de la voluntad del sujeto declarante, y de lo que le interese o considere oportuno plasmar en esa declaración.
4. COMPARACIÓN METODOLÓGICA CON LA FDA
4.1. Creación y evolución
Conviene comenzar indicando que la Foods and Drugs Administration es la agencia federal más antigua dependiente del Gobierno de los Estados Unidos. Comenzó su labor en 1848 cuando la tarea principal que desempeñaba se basaba en la monitorización de los productos químicos utilizados en la producción agrícola, esta función después pasó a depender del Departamento de Agricultura, que se la asignó específicamente a la FDA. Aunque su denominación actual data de 1930, sus atribuciones regulatorias se basan en la Pure Food and Drugs de 1906, que le otorgaba la competencia para restringir el comercio interestatal cuando se despertasen dudas respecto de la calidad y pureza de determinados productos, también farmacéuticos.
En 1933 se promovió una legislación ante el Senado de Estados Unidos para sustituir la Ley de 1905 a consecuencia de la catástrofe producida por la empresa Massengill Co., en Bristol —Tennessee— al incorporar congelante de automóvil en un elixir para uso humano, con el resultado de 107 personas fallecidas. De esta forma, en 1938 se aprobó por el Congreso la Federal Food, Drug, and Cosmetic Act (FD&C Act), conforme a la cual se exigía la obligación a las empresas que pretendiesen comercializar un medicamento que garantizasen su seguridad. De este modo, antes de poder poner en el mercado una drug —medicamento— quedaban obligados a presentar una solicitud de aprobación ante la FDA, partiéndose de la premisa del silencio positivo, es decir, si transcurrido un plazo no había respuesta por parte de la Agencia, el producto quedaba aprobado.
En octubre de 1962 se aprobaron las denominadas Kefauver-Harris Drug Amendments respecto de la Federal FD&C Act, por las que las compañías tenían que probar no sólo la seguridad del medicamento sino también aportar evidencia contundente respecto a la eficacia del mismo respecto al objetivo perseguido, de este modo se introdujo el revolucionario requisito —para la época— de la aportación de estudios científicos solventes y documentados. Al mismo tiempo, estas enmiendas implicaron la obligatoriedad de presentar una solicitud de autorización de comercialización ante la FDA antes de proceder a su puesta en el mercado.
Por su parte, en 1966 se aprobó la Fair Packaging and Labeling Act que exigió unos requisitos reforzados de etiquetado y de empaquetado respecto de los medicamentos dirigidos al uso humano, y también consecuencia de una catástrofe sanitaria derivada del envoltorio de ciertos medicamentos.
A su vez, en 1981 se fortalecieron los criterios atinentes a la protección de las personas en relación con ciertos medicamentos, y se mejoraron los Institutional Review Boards (IRBs), que son los paneles de científicos y especialistas que aseguran, tanto desde los hospitales como desde los centros de investigación, la seguridad y el bienestar de los pacientes sujetos a pruebas clínicas derivadas del desarrollo de determinados medicamentos o nuevas sustancias.
Del mismo modo, queda bajo la jurisdicción de la FDA la comprobación de los requisitos para el consentimiento informado respecto de las personas físicas que voluntariamente se sometan a ensayos clínicos relacionados con procedimientos de aprobación de medicamentos. En este sentido, los drugs sponsors —los promotores de medicamentos— tienen que aportar a la FDA los resultados de los estudios previos llevados a cabo sobre animales, para que se puedan autorizar las pruebas respecto de humanos. Toda esta información se presenta en la denominada IND o INDA —investigational new drug application—, que se somete a la revisión tanto de la FDA como de los IRB —Insittutional Review Boards—.
Respecto de las enfermedades raras y su tratamiento, en 1983 se aprobó la Orphan Drug Act —ODA—, que alentaba la investigación y estudio de nuevos medicamentos relacionados con el tratamiento de las mismas —que con anterioridad quedaban excluidas de tratamiento efectivo por la falta de interés de las empresas farmacéuticas en su estudio, y esto debido a las escasas perspectivas de beneficio económico que podían conllevar—. Esta legislación promovió incentivos fiscales, créditos y financiación de investigaciones clínicas, incluyendo una extensión temporal en la exclusividad de comercialización para la empresa farmacéutica que obtuviese la primera aprobación de la FDA respecto de un medicamento relacionado con una enfermedad rara.
En 1992 se aprobó la Prescription Drug User Fee Act —PDUFA— que introducía un sistema de pago de tasas y derechos a la FDA por parte de las empresas farmacéuticas, de manera que esta contribución financiera permitió aumentar su staff en un 60 por cien, y reducir el plazo de tramitación de las solicitudes de autorización para medicamentos a diez meses. A su vez, desde 1997, la Food and Drug Modernization Act —FDAMA— ha implicado una aceleración todavía mayor en la tramitación de las solicitudes, otorgando al mismo tiempo una exclusividad de comercialización de seis meses suplementarios para aquellos desarrolladores que lleven a cabo estudios también relacionados con ciertas enfermedades infantiles.
4.2. Organización y estructura
La FDA es una agencia de carácter federal, dependiente del Department of Health and Human Services, formada a su vez por nueve órganos, sitos en trece sedes oficiales, simplemente van a ser enumerados, aunque uno de ellos será estudiado con mayor profundidad: Center for Biologics Evaluation and Research, Center for Devices and Radiological Health, Center for Drug Evaluation and Research, Center for Food Safety and Applied Nutrition, Center for Tobacco Products, Center for Veterinary Medicine, National Center for Toxicological Research, Office of Regulatory Affairs, Office of Operations.
Cada uno de esto órganos tiene una estructura propia a cuya cabeza está el Commissioner de la FDA, que dispone de su propia Oficina.
La oficina encargada del estudio y aprobación de las solicitudes de autorización de medicamentos es el Center for Drug Evaluation and Research (CDER) 111, siendo hasta cierto punto el equivalente del Comité para Medicamentos de Uso Humano en la EMA, de manera que es el órgano dentro de la FDA que articula los estudios y análisis científicos —Drug Efficacy Studies (DES)— que deben acompañar a una NDA para su aprobación, y emite la decisión definitiva respecto al medicamento sometido a consideración..
En relación a los medicamentos que se aprueban anualmente por la FDA y la EMA, se pueden distinguir dos categorías de medicamentos, los genéricos y los nuevos medicamentos. Respecto a la primera categoría, la diferencia cuantitativa de autorizaciones de comercialización a favor de la FDA es abrumadora; sin embargo, respecto a los medicamentos de nuevo cuño, la diferencia —aunque también sea favorable a la FDA para determinadas categorías (por ejemplo, tratamientos de cáncer y hematología)— refleja un mayor equilibrio entre ambas agencias.
4.3. Resultados comparativos
De este modo, estudios solventes 112 reflejan que, respecto a aprobaciones de comercialización en relación con genéricos: en el año 2017, la EMA aprobó 22, la FDA 843; respecto del 2018, la EMA aprobó 9, la FDA 810; en 2019, 15 medicamentos por la EMA, 836 la FDA; en 2020, 61 por la EMA, 3.243 por la FDA: es decir, en el tramo temporal comprendido entre 2017 y 2020, la EMA autorizó 61 genéricos, mientras que la FDA aprobó 3.243. De hecho, el informe citado explica como los diferentes procedimientos y requisitos de aprobación entre ambas agencias provoca la situación singular que se manifiesta en que hay determinados medicamentos que se aprobaron hasta con 17 años de antelación en la FDA respecto de la autorización concedida en la EMA 113.
Sin embargo, también encontramos evidencia científica que afirma una cierta concordancia en los resultados de los procesos de aprobación respecto de solicitudes de comercialización en ambas agencias 114: de hecho, hay estudios que reducen esa diferencia sustancialmente, siendo que la initial review podría necesitar de media 303 días en la FDA, mientras que en la EMA ascendería 366, y para las full reviews este espacio temporal se podría elevar de 322 días en la FDA a 366 en la EMA —debido a la no diferenciación entre aprobación definitiva y provisional en la EMA— 115.
En este sentido, en el Informe anual que el CDER publica respecto de los nuevos medicamentos aprobados, en el año 2020 116, este órgano aprobó 53 nuevos medicamentos (frente a 39 aprobados en la EMA), además el informe expresa como 21 de entre ellos fueron clasificados first in class, es decir, que se engloban dentro de los medicamentos que tienen unos mecanismos de acción distintos a los que se encontraban comercializados hasta entonces, de manera que tienen un gran potencial de mejora en las terapias en que se utilicen. Otro hecho relevante y que la significa frente a la Agencia europea, es que el 58 por cien de los fármacos autorizados son huérfanos, es decir, están indicados para el tratamiento de enfermedades raras. Asimismo, resulta interesante indicar que el 68 por cien de los medicamentos se aprobaron por alguno de los sistemas de fast track, es decir, de aprobación rápida.
Respecto a la puesta a disposición de los fármacos respecto del paciente —el consumidor final de los mismos—, la evidencia demuestra que en Estados Unidos se alcanza esta fase con una antelación media de 90 días respecto de la europea, y 355 días antes que en Canadá. Esta situación es particularmente significativa en medicamentos relacionados con el tratamiento del cáncer, donde los intervalos de puesta a disposición al paciente siempre son menores en Estados Unidos que en Europa, y esto es así respecto de todos los medicamentos considerados 117.
También hay diferencias relevantes a efectos de la publicación y transparencia respecto de la comunicación de los resultados de las evaluaciones y documentos anexos relevantes: en la FDA se publican a posteriori la práctica totalidad de los resultados de la evaluación científica —con independencia de que tengan trascendencia o relevancia comercial 118-
En la EMA la comunicación de los resultados de una evaluación toma en cuenta, como uno de los criterios restrictivos respecto de la misma —como ya se ha explicado— la consideración o calificación de información commercially sensitive —sensible comercialmente—, y sólo se publican cuando prevalece un interés público superior 119.
En la FDA existen cinco tipos de solicitudes de aprobación: La Investigational New Drug Application (IND) que se refiere a la fase previa de examen, estudio y valoración de los resultados farmacológicos del fármaco; la New Drug Application (NDA) que recoge el conjunto de la evidencia aportada en el estudio previo realizado por el CDER, y que expresa la solicitud formal completa; la Abbreviated New Drug Application (ANDA), que se reserva para las solicitudes de aprobación de genéricos; la Therapeutic Biologics Application (BLA) para inmunoterapias que no supongan vacunas; y la Drug Applications for Over-the-Counter (BLA), estos fármacos no necesitan prescripción médica, y se pueden comprar sin receta, de ahí que se les califique de OTC.
De esta forma, nos encontramos en la FDA con un sistema de solicitud de autorización que al menos se puede calificar de dual, en la medida en que distingue entre la fase de investigación inicial —IND (Investigational New Drug)—, que será sometida a la supervisión de un IRB —Institutional Review Board—; y la NDA —New Drug Application— en sí misma considerada, que es la formal y definitiva solicitud de autorización, que engloba todos los ítems recopilados y contrastados en la fase de investigación y asistencia científica, y sometida a ensayos clínicos y que recibirá la eventual aprobación de la FDA.
De esta forma, y conforme al esquema expuesto, la IND permite llevar a cabo pruebas del medicamento por parte de los investigadores que lo estén desarrollando, de manera que los resultados pasarán a integrar esa solicitud formal de autorización denominada NDA. De este modo, en Estados Unidos, la presentación de una IND garantiza que un IRB deberá estudiar exhaustivamente la evidencia científica para así aprobar los resultados de los estudios preliminares de ese medicamento, antes de que se siga adelante con el proceso de aprobación por el CDER.
Otra interesante cuestión que se suscita en la consideración de las diferencias entre la EMA y la FDA hace referencia al nivel de implicación del staff de una y otra en los procedimientos de aprobación. En la FDA tradicionalmente se han suscitado cuestiones relativas a la salvaguarda de la independencia de la Agencia, de manera que se temía que una
Too much participation by FDA staff in the development process would leave the Agency unable to be properly neutral and analytical when resulting data were submitted as part of an NDA 120
Esta es una consideración que ya hemos plasmado en el presente trabajo respecto de las consecuencias de la excesiva implicación de la EMA en la asistencia científica previa a la valoración de la solicitud. Entendemos que esta es una inquietud que se siente de manera más aguda en la FDA 121, y que se expresa en el capítulo dedicado a Standards of Conduct and Conflicts of Interest dentro del Code of Federal Regulations 122, que recoge los conceptos y situaciones de las que se debe inferir que hay un conflicto de interés, con el sometimiento de las mismas a un ente autónomo dentro de la FDA —cuyos miembros dependen directamente del Comisionado de la Agencia—, y que se denomina Conflict of Interest Review Board, que se compone de cinco miembros, y que puede ser apelado por cualquiera que ostente un interés legítimo en la resolución recaída, o en la resolución de ese conflicto de interés 123.
Es importante recordar que en la EMA no existe un órgano o ente equiparable, y esta labor —eventualmente— se llevaría a cabo por el Comité de Medicamentos para Uso Humano.
A continuación, incluimos un gráfico que expresa las diferencias en el tratamiento de las solicitudes de autorización entre la FDA y la EMA, haciendo referencia a las causas de esa distinción y el número de solicitudes que estaban afectadas:
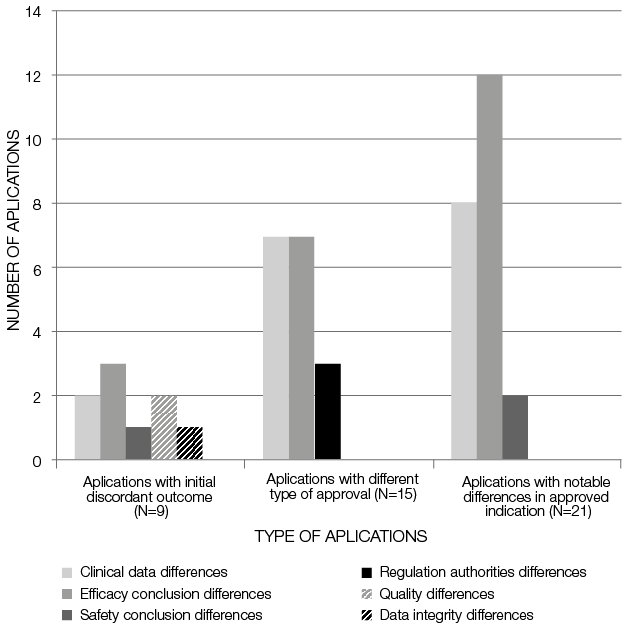
De hecho, el gráfico sirve para constatar los distintos criterios utilizados en las agencias para fundamentar decisiones distintas, siendo el más interesante el tercer grupo —el referido a solicitudes con diferencias notables respecto a las indicaciones aprobadas —applications with notable differences approved indication (N 21)— que respecto de 21 solicitudes similares constata que hay diferencias notables en los ensayos clínicos respecto de 8 solicitudes, y también en relación a las conclusiones sobre su eficacia —en 12 del total— .
A pesar de todo, y a modo de ejemplo, se puede derivar una cierta concordancia en la tasa de aprobación de solicitudes en la EMA y en la FDA para el espacio temporal comprendido entre 2014-2016: de este modo, respecto de 107 medicamentos, la FDA aprobó 91, denegó 14 solicitudes, y 2 fueron retiradas; respecto a los mismos medicamentos, en ese tramo temporal, la EMA aprobó 90 medicamentos, rechazó 3, siendo retiradas 6 solicitudes. De esta forma, el resultado regulatorio es muy similar, con una concordancia del 92 por cien, y siendo la discordancia de un 8 por cien respecto de 9 solicitudes respecto de 107 sometidas a examen.
Por su parte, los plazos de tramitación de las solicitudes de autorización son sustancialmente inferiores de media en la FDA que en la EMA —pudiendo reducirse a menos de la mitad los plazos entre una y otra, ya que en la EMA superan de media los dos años, mientras que en la FDA está en el orden de los seis meses, dependiendo del tipo y alcance del medicamento—.
El siguiente cuadro también refleja cierta concordancia en los porcentajes de aprobación respecto de 98 solicitudes de medicamentos similares ante la FDA y la EMA, siendo ligeramente superior respecto de ésta última en lo que hace referencia a las aprobaciones estándar —84 en la EMA, frente a 79 en la FDA—, mientras que la FDA aprobó 19 a través de la vía de la aprobación acelerada, mientras que la EMA otorgó 11 aprobaciones condicionales 124.
|
EMA marketing approval type, N = 98 |
||||
|
Standard (84) |
Conditional (11) |
Exceptional (3) |
||
|
FDA |
Standard |
74 (76%) |
2 (2%) |
3 (3%) |
|
Accelerated approval (19) |
10 (10%) |
9 (9%) |
N/A |
|
Los procedimientos de aprobación acelerada ante la FDA se refirieron a medicamentos oncológicos en 12 casos, y relacionados con hematología en 5 ocasiones, mientras que en la EMA las autorizaciones bajo circunstancias excepcionales se refirieron únicamente en 7 supuestos a medicamentos oncológicos, y en 2 a medicamentos relacionados con hematología.
Es evidente que resulta complejo aplicar baremos uniformes a las dos agencias, ya que van a depender tanto del período temporal considerado, como de los criterios que se apliquen para la consideración de los procedimientos utilizados tanto en una como otra institución, de modo que las concordancias quedan muy relativizadas según recurramos a un tipo de parámetro u otro. Este trabajo no pretende ser en absoluto un estudio médico-científico, ya que excedería el objeto del mismo y lo desnaturalizaría, pero sí resulta conveniente introducir algunas conclusiones en relación con los datos expuestos.
Sí resulta importante señalar que una de las diferencias fundamentales entre la EMA y la FDA se manifiesta en la existencia en ésta de mecanismos de aprobación acelerada, que se reservan particularmente respecto a fármacos que puedan ser eficaces para enfermedades incurables o de tratamiento desconocido. Mientras que en la FDA nos encontramos con tres procedimientos distintos de aprobación de solicitudes de comercialización a través de una vía rápida 125: Fast Track, Breakthrough Therapy y Priority Review, la EMA dispone también de mecanismos de urgencia, pero no son tan flexibles como los americanos de la FDA.
La FDA en 2019 autorizó alrededor del 60 por cien de las solicitudes que se le plantearon a través de algún sistema de aprobación rápida, lo que la distingue claramente de las autorizaciones emitidas por la EMA a través de sus procedimientos equivalentes. Como crítica frente a las técnicas de aprobación con mayor celeridad, se alega —respecto de la FDA— que supone un ingreso económico superior para su presupuesto, debido al mayor número de medicamentos aprobados, lo que puede también suponer un incremento del peligro respecto a la seguridad final del medicamento o sustancia, algo que afecta de forma negativa a los receptores finales del medicamento.
Por su parte, la EMA consta de procedimientos de aprobación más lentos y tediosos, que quizá —eventualmente— hacen prevalecer otro tipo de criterios, aunque su crítica fundamental es el retraso superior en poner en disposición del enfermo terapias y medicamentos que, sin embargo, en Estados Unidos son disponibles de manera más veloz.
Es evidente que no es fácil llegar a un compromiso entre los elementos puestos de manifiesto, ni resulta conveniente llevar a cabo declaraciones maximalistas relativas a la mayor o menor bondad de los procedimientos seguidos en una y otra agencia, pero es innegable que la pandemia ha puesto en evidencia las consecuencias de la distinta metodología utilizada por la EMA y la FDA, especialmente en relación con la puesta a disposición del público de tratamientos eficaces contra la COVID 19 126.
Si se considera única y exclusivamente la perspectiva garantista, la EMA puede resultar favorecida, aunque quizá para determinados tratamientos y terapias —especialmente respecto de medicamentos innovadores frente a enfermedades sin tratamiento eficaz como pueden ser el cáncer— la celeridad que introduce la FDA puede constituir un factor relevante y extremadamente importante, porque está en juego la eventual expectativa vital del receptor del mismo, es decir, del paciente. Este criterio no debería obviarse a la hora de ponderar las ventajas de una u otra estrategia de autorización.
5. PROPUESTAS DE MEJORA REGULATORIA DE LA EMA
No resulta sencillo elaborar o propugnar propuestas de mejora de procedimientos que están explicitados en normas de carácter general, y que han sido contrastados a través de un práctica constante y reiterada durante muchos años de actividad —como en el presente supuesto concurre en el caso de la Agencia Europea de Medicamentos—, pudiendo ser fácil incurrir en generalidades y trivialidades exentas de fundamento. Esto resulta más complejo aún si se quiere evitar llevar a cabo una crítica superficial o insustancial.
Sin embargo, nos vemos en la obligación de enunciar y suscitar algunos puntos que se nos han presentado a lo largo de estas páginas, y que —de nuevo, conviene indicarlo— buscan un acercamiento positivo y constructivo a la importante labor llevada a cabo por la Agencia, pretendiendo resaltar cuestiones que podrían reconsiderarse, ponderarse, e incluirse en un eventual Libro Blanco que pudiese hipotéticamente revisar algunos de los métodos y procesos seguidos por este importante organismo. Pasaremos a enumerar algunos de los que, reitero, se nos han presentado subjetivamente como dignos de reflexión.
5.1. Criterios para la elección de los miembros de los órganos
La Agencia Europea de Medicamentos tiene una naturaleza mixta, que la dota de una cierta ambigüedad jurídica, en la medida en que como agencia descentralizada forma parte de la estructura organizativa y orgánica del esquema institucional de la Unión Europea, y más concretamente, de la estructura de la Comisión Europea, al ser ésta la Institución que centraliza la mayoría de los órganos y entes descentralizados —ya sean de naturaleza ejecutiva o consultiva—.
De este modo, esa naturaleza le otorga a la EMA un amplio margen de autodeterminación y organización, aunque su actividad queda permeada por algunos de los elementos que ya en la Comisión Europea se perciben en su actividad ordinaria: por un lado, la EMA tiene un importante elemento nacional, al ser sus miembros propuestos por los Estados miembros, de manera que tanto el Consejo de Administración como los Comités más importantes —el de medicamentos para uso humano, y el de medicamentos para uso veterinario— tienen un representante de cada país, que también nombra un suplente.
Esta situación inequívocamente permite que el componente político esté presente en la acción de la Agencia: es indudable que este es un factor que se compadece mal con las exigencias derivadas de la objetividad, y también de la independencia a la hora de juzgar y examinar con parámetros ajenos a la exclusivamente científica. Por esta razón, en una revisión del Estatuto jurídico de la EMA podría plantearse una disminución del componente nacional, para evitar situaciones que sutilmente pudiesen influir en sus consideraciones, imbuyéndolas de elementos exorbitantes a los exclusivamente científicos. La FDA no tiene ningún parámetro territorial en su configuración —siendo una agencia federal en la que también, eventualmente, los Estados podrían pretender una representación—, algo que podría servir de criterio para una eventual revisión de la Agencia europea.
5.2. Separación de las funciones de asesoría científica y emisión de opinión
Por otra parte, la Agencia Europea de Medicamentos manifiesta una contradicción en sus funciones, que se manifiesta en la forma de actuar sus competencias, en la medida en que tiene una naturaleza mixta que —en nuestra opinión (que en absoluto es científica o técnica, sino únicamente jurídica)— puede provocar conflictos de interés relevantes: la EMA es órgano regulador, con naturaleza decisoria indirecta —recordemos que propone la autorización a la Comisión Europea de medicamentos que se van a comercializar en los Estados de la UE—, y también es órgano asesor, en la medida en que se implica en la asesoría científica previa a la autorización del principio o medicamento en cuestión.
Esta confusión o ambigüedad, además, puede tener repercusiones en las decisiones finales que se tomen por parte de la Agencia: por un lado, hay solicitantes que pueden prescindir del apoyo científico de la Agencia —algo que como demuestran las estadísticas, objetivamente reduce las posibilidades de concesión de la autorización, factor que en absoluto es irrelevante a la hora de afrontar una estrategia de autorización y comercialización por parte de una compañía farmacéutica—; de otra parte, el recurso a la comitology 127—es decir, a grupos o comités de expertos organizados ad hoc— es algo que ya se ha puesto de manifiesto reiteradamente en la Comisión Europea, con una continua apelación a esta técnica, lo que tradicionalmente ha provocado complejas situaciones de transparencia, de falta de rendición de cuentas, y de eventuales conflictos de interés, siendo uno de los elementos identificados en todas las propuestas de mejora regulatoria respecto de esta Institución europea 128.
La realidad es que los grupos de expertos, su formación, la determinación de su composición, no son elementos irrelevantes respecto a la propuesta de autorización de comercialización, y si a esto le añadimos su eventual capacidad de suscitar situaciones relativas a la consideración de su independencia —no sólo respecto del Estado que les proponga, sino también de eventuales centros de investigación, universidades, empresas farmacéuticas u otros actores implicados en el mundo del desarrollo de medicamentos—, nos encontramos con situaciones de difícil gestión, y que pueden afectar a la actividad, prestigio y capacidad de independencia de la Agencia.
En este sentido, se puede citar como ejemplo claro de conflicto de interés el asunto suscitado por la empresa Pharma Mar S.A., en el que se ventiló su pretensión de anulación de una decisión de denegación de autorización de comercialización de un medicamento emitida por la Comisión Europea, y sustanciada en el Asunto T-598/18.
Phama Mar puso de manifiesto en sus alegatos en el recurso ante el Tribunal General que se había lesionado una serie de derechos básicos: en concreto, entendió que no se había garantizado la imparcialidad en el Grupo asesor de oncología, debido a la contaminación de dos de sus miembros; también se suscitó la transgresión de derechos derivados de la buena administración y de igualdad de trato; por último, se alegó que se habían atacado los derechos de defensa y la obligación de fundamentar las decisiones.
El Tribunal General llega a la conclusión en su sentencia de que la composición del SAG no era imparcial, al incurrir dos de sus componentes en causas de incompatibilidad objetiva, entre otros motivos, por su relación con un hospital universitario que cuenta con un centro de investigación que participa en el desarrollo y producción de un medicamento equivalente en competencia con el desarrollado por la empresa española.
Estas circunstancias nos permiten ejemplificar la dificultad que implica la ponderación de las causas de incompatibilidad —distinguiendo el TJUE entre imparcialidad objetiva y subjetiva—, y los conflictos de interés subyacentes a las mismas. La Sentencia de 28 de octubre de 2020 anuló la decisión de la Comisión Europea —que había sostenido la legalidad del procedimiento—, y la irrelevancia del papel del SAG —Scientific Advisory Group— en la propuesta de decisión del CMHP, y, por lo tanto, sobre la decisión final contestada por la empresa española.
La Sentencia del Tribunal General fue recurrida por dos Estados miembros conforme a la legitimación que ostentan de acuerdo a los Tratados —Estonia y la República Federal de Alemania—, y no por la propia Comisión Europea —siendo ésta quien, en primer término, tendría un mayor interés en aclarar la legalidad de la denegación de solicitud que había dictado conforme a la propuesta de resolución emanada desde la EMA—. El Tribunal de Justicia recientemente ha dictado Sentencia en el recurso planteado 129 conforme a la cual se devuelve el asunto al Tribunal General. Entiende el Tribunal de Justicia que “el legislador de la Unión habilitó a la EMA para llevar a cabo una ponderación entre, por un lado, la doble exigencia de imparcialidad y de independencia de sus expertos y, por otro, el interés público en disponer del mejor asesoramiento científico posible sobre cualquier cuestión relacionada con la evaluación de la calidad, la seguridad y la eficacia de los medicamentos de uso humano” 130. De esta forma, el Tribunal en el recurso entiende que se produjo un error de Derecho por parte del Tribunal General “al considerar que el hospital universitario en cuestión constituía una <compañía farmacéutica> por el mero hecho de que se controlaba un centro de terapia celular que reunía, él mismo, los criterios de una <compañía farmacéutica> 131.
De esta forma, se puede constatar a través de este caso concreto la complejidad que despliega la ponderación, baremación y consideración de la independencia de los miembros de los comités científicos en el seno de la EMA, y la dificultad que, incluso en sede judicial, entraña desligar las vinculaciones de los distintos expertos y científicos que prestan sus servicios en la fase de asesoría científica previa a la solicitud de autorización.
Todo ello nos lleva a reiterar que esta confusión de roles en la Agencia sólo puede limitarse a través de una reconsideración profunda de su naturaleza, de sus funciones, y, por lo tanto, del sistema de asesoría científica, que se acepta como un a priori indubitado en el esquema vigente, cuando a nosotros no se nos antoja como tal. En esta línea, sería clarificador que se pudiese contar con una disposición jurídica similar a la que está vigente respecto de la Comitología en la Comisión Europea 132, creando un registro público de expertos equivalente para la EMA (el equivalente al Comitology Register 133), y que puede contribuir sustancialmente a mejorar el acceso al expertise que maneja la Agencia, aportando transparencia al conjunto del proceso, y mejorando su resultado final.
5.3. Clarificación y simplificación de la estructura orgánica
Otra cuestión que se nos ha ido suscitando a lo largo del trabajo ha sido la difícil imputación de responsabilidades y riesgos: la Agencia Europea de Medicamentos consta de un complejo aparato administrativo, con una pléyade de comités y órganos con siglas y acrónimos de difícil comprensión —SAWP, CAT, COMP, RMP, PRAC, SmPC, Enpr-EMA, ENCePP, PDCO, CHMP, EPAR, Clock Stop, etc.—, lo que respecto del profano no contribuye en absoluto a una sencilla comprensión del esquema de funcionamiento interno de la misma. De nuevo, la apelación reiterada y continuada a esta técnica no fomenta la transparencia, no aporta a la simplificación administrativa, y tampoco colabora a la mejora regulatoria 134, como principio director y guía en la acción de las Instituciones Europeas y de sus órganos y agencias descentralizados, y que se manifiesta en la Smart Regulation 135. Además, un esfuerzo más simple contribuiría a otorgar claridad al sistema, ya de por sí complejo y poco asequible a efectos no sólo del propio solicitante sino también del experto en Derecho de la Unión.
5.4. Reforma del actual enfoque formalista de la transparencia
En la Agencia Europea de Medicamentos se ha adoptado un enfoque formalista de la transparencia, con esto queremos indicar la prevalencia de una concepción basada en la información aportada por el declarante, conforme a un sistema de manifestación de los inputs de información que se encuentran a su vez baremados y prefijados. La EMA juzga las posibles situaciones de conflicto de interés y les atribuye un nivel de riesgo que permite decidir respecto de qué actuaciones le quedarán vetadas al incurso en la incompatibilidad. De esta forma se parte de una perspectiva formalista que puede generar conflictos de interés respecto de los nombramientos de expertos y asesores.
Entendemos que, en primer lugar, no es una técnica que parta de la premisa de compartir la información con los sujetos eventualmente afectados —es decir, con el solicitante de la autorización de comercialización—, que, de esta forma, queda al albur de la Agencia respecto a la decisión de la participación del experto o miembro que incurra en la incompatibilidad. En nuestra opinión, la solución de excluir nominalmente —por ejemplo— del voto final a un experto que sí ha podido influir de forma efectiva en la deliberación —en la que tome parte con la presentación de su informe— no resulta ni suficiente ni admisible, y puede influir de forma gravosa en la tendencia de voto respecto de los que sí puedan ejercitarlo en el Comité o Grupo correspondiente.
Además, resulta importante reseñar como el Tribunal de Justicia de la UE se ha manifestado de manera contundente respecto a las condiciones de comunicación de información confidencial respecto de terceros 136: de manera que reconociendo que se puede partir de una presunción general de confidencialidad, ésta puede ceder en la medida en que la Agencia, órgano o Institución comunitaria entienda que su comunicación puede llevarse a cabo por no concurrir ninguna de las excepciones que se recogen en el Reglamento 1049/2001 —es decir, las que se expresan en el artículo 4.2 del mismo 137—.
De este modo, de nuevo, en la EMA recae la responsabilidad última respecto de la publicación de la información confidencial que conste en su poder. A sensu contrario, también se puede entender —conforme a lo expuesto por el TJUE— que cuando circunstancias particulares del solicitante exijan esa discreción, debería ponderarse muy detenidamente la conveniencia del acceso de terceros a la información sensible desde la perspectiva comercial 138.
Asimismo, también se pone de manifiesto esta tendencia a la transparencia como mero criterio formalista en los criterios de publicación de agendas y de minutas de reuniones, tanto de Comités y Grupos de trabajo, como del Consejo de Administración. El mero recurso a la publicidad de los resultados de la votación final, sin saber los votos divergentes y quién los emitió, o —respecto de los temas tratados— simplemente hacer público el denominado high level procedure outcome or milestone 139—es decir, aquellas cuestiones que la Agencia entienda más relevantes o significativas de las tratadas en cada sesión— resulta claramente insuficiente e insatisfactorio.
También se percibe este enfoque en el criterio para la publicación de los votos divergentes, que sólo se lleva a cabo a posteriori en los supuestos en que se haya solicitado un re-examen respecto de la decisión recaída 140, y por lo tanto, cuando ya se ha producido una toma de posición.
Como propuesta indicamos la conveniencia de adoptar un enfoque más pragmático de la transparencia, que no quede encorsetada en meros parámetros prefijados, resultado de la declaración subjetiva en un formulario previo, ateniéndose más a criterios desarrollados desde la propia EMA, que respondan a la práctica y experiencia desarrollada desde la Agencia, dotándola incluso de un órgano interno específico que se ocupe exclusivamente del análisis y ponderación de los riesgos que puede suponer la participación de un determinado experto o miembro de un comité en cualquier fase de la aprobación de una solicitud de comercialización. Esta técnica es la que se asume por la FDA a través del panel que revisa los conflictos de interés 141. Además, esto sólo puede redundar en el prestigio de la institución, y contribuir a disminuir los riesgos inherentes a esa dual perspectiva que se da cita en la EMA —asistencia científica y órgano decisor— que la hace vulnerable a este tipo de situaciones.
En este sentido, se puede hacer referencia al clásico debate de reglas contra principios, iniciado por Dworkin y Hart a principios del siglo pasado, pero todavía de actualidad 142. Existen en Derecho comparado dos concepciones opuestas sobre las obligaciones jurídicas: una defiende que las obligaciones se especifiquen en reglas concretas para garantizar la seguridad jurídica, mientras que la otra critica el excesivo formalismo de un sistema de reglas y considera necesarios los principios generales del Derecho.
El ordenamiento americano ha sido tradicionalmente defensor de la primera postura, mientras que el Derecho de la Unión Europea se caracteriza por favorecer los principios. Así, la situación de la EMA es atípica en el entramado europeo en tanto que ha optado por diseñar un sistema de garantías de transparencia totalmente rígido, compuesto por multitud de normas, que da pie a que se transgreda el sentido de la ley aun respetando la letra de la misma.
Por esta razón, creemos que una manera de reducir la formalidad excesiva de esta normativa sería introducir y desarrollar baremos y criterios ínsitos en los principios generales reconocidos en el Derecho de la Unión, como el principio de imparcialidad o el de interdicción de la arbitrariedad, en la regulación de la EMA. En lugar de limitarse las obligaciones al tenor literal de una regla rígida de compleja interpretación, se podría exigir para cada decisión de la EMA un razonamiento fundado sobre cómo se han respetado en ese caso concreto los principios aplicables; siendo así más sencillo evaluar la actuación de la EMA, de sus miembros y expertos, y así —eventualmente— poder más fácilmente exigir responsabilidades.
5.5. Refuerzo del derecho a la defensa
La práctica cotidiana de la Agencia pone de manifiesto situaciones que objetivamente denotan circunstancias y elementos de mejora, que no son irrelevantes, y que —de nuevo— dejan en evidencia flaquezas y debilidades que repercuten de manera perjudicial sobre los particulares y empresas que solicitan autorizaciones. La EMA es órgano de naturaleza pública, sometida a los principios de Derecho administrativo europeo, y que —como hemos puesto de manifiesto— queda sujeta a varios códigos de buena conducta administrativa. Estos códigos otorgan derechos subjetivos que acrecen al patrimonio jurídico de particulares y agentes económicos, y que pueden ser alegados frente a la Agencia, y que no se pueden obviar, ignorar o denostar con meras e infundadas apelaciones al bien público o al interés general.
Por supuesto, es evidente que la EMA está vinculada por los principios reconocidos en los Tratados europeos —es decir, en el Derecho Originario de la UE—, y, en concreto, por los derechos fundamentales que se reconocen en aquellos: nos estamos refiriendo a la tutela y respeto del derecho de defensa, que forma parte de ese citado patrimonio jurídico del Derecho de la Unión Europea, reconocido por la Carta de Derechos Fundamentales de la Unión Europea en su artículo 48 143. El derecho a la defensa es expresión básica del concepto de ciudadanía, y es predicable tanto de personas físicas como jurídicas; la práctica cotidiana de la Agencia pone de manifiesto tendencias, actitudes y prácticas consolidadas que atentan gravemente contra este derecho fundamental, pudiendo llegar a poner en entredicho la credibilidad del conjunto del sistema de autorización de medicamentos en la UE. De esta forma, cuando la EMA presenta una opinión desfavorable contradiciendo el criterio previo de los rapporteurs lo que se lesiona es el derecho de defensa del solicitante, que obliga a que éste tenga la posibilidad real y efectiva de participar en el proceso con garantías de contradicción y de igualdad de armas, que son presupuestos consustanciales e irrenunciables al contenido esencial del expresado derecho fundamental.
De nuevo, vamos a continuar con la exposición del caso citado con anterioridad para ejemplificar las consideraciones que hemos expuesto, y en el que se ha puesto de manifiesto una eventual lesión al derecho de defensa, con todas las perjudiciales consecuencias que conlleva para las pretensiones del demandante: como vimos, los hechos se sustanciaron ante el Tribunal General de la Unión Europea en el asunto T-594/18 144, que sustanció la pretensión de anulación de una decisión de la Comisión Europea denegando una autorización de comercialización por conflictos de interés dentro del CMUH 145, y que fue suscitado por parte de la empresa española PharmaMar SA.
En este caso, se emitieron en el proceso de autorización cuatro opiniones por dos rapporteurs y co-rapporteurs distintos, ya que el solicitante de la autorización —la empresa PharmaMar— pidió la revisión del primer dictamen. En consecuencia, la decisión de la EMA fue emitida pese a la opinión favorable de los primeros rapporteur y co-rapporteur, que sí se mostraron unánimemente a favor de la comercialización. Sin embargo, en la segunda opinión se perdió esa unanimidad al cambiar al rapporteur y al co-rapporteur, ya que se emitieron conclusiones divergentes.
En este sentido, se puede afirmar que el hecho de que la opinión de la EMA se aparte de la opinión unánime de los primeros rapporteur y co-rapporteur puede quizás calificarse como una situación extraordinaria, pero no deja de ser una opción legítima que necesariamente ha de tener cabida en un sistema como el de la aprobación de medicamentos que está compuesto por varias fases en las que intervienen expertos independientes.
Sin embargo, ante una situación extraordinaria como es el apartarse de la opinión unánime de relator y co-relator, debería existir es un procedimiento especial para garantizar el derecho de defensa de los solicitantes y evitar que cambios tan radicales en la valoración de la evidencia científica se realicen sin transparencia y sin asegurar al solicitante una genuina oportunidad de volver a discutir la evidencia científica con los relatores.
5.6. Agilización de los trámites
Para culminar este trabajo, nos gustaría indicar que convendría —en coherencia con todo lo expuesto anteriormente— que la EMA adoptase procedimientos más veloces en relación con la tramitación, estudio y eventual resolución final de las solicitudes que se le plantean de autorización de comercialización. La FDA aprueba con mayor celeridad, de manera más eficiente, y con resultados más ventajosos para todas las partes implicadas, comenzando por el paciente final del fármaco, siendo éste quién más clara y unívocamente encarna el bien común, y también ese diluido interés general, cuya vacua invocación tantas veces puede eventualmente ocultar otro tipo de intereses.
CONCLUSIÓN
1. La Agencia Europea de Medicamentos es fundamental en la realización de la competencia atribuida a las Instituciones Europeas de salvaguardar la salud pública, como manifestación del bien común. Sus funciones, estatuto y prerrogativas la sitúan en una posición singular respecto a otras Agencias descentralizadas europeas, y conviene someter su régimen a una mejora continua, para que pueda responder eficazmente a los retos con los que se enfrenta.
2. La EMA tiene una naturaleza dual, en la medida en que presta asesoría científica a los promotores de fármacos —implicándose de esta forma en el desarrollo del compuesto—, y por otra, lleva a cabo una propuesta de resolución respecto de la autorización de comercialización del mismo: este sistema mixto puede suscitar problemas de transparencia, conflictos de interés, falta de imparcialidad e independencia, cuya gestión es trascendental para la aprobación o denegación de la autorización, y que necesitan ser afrontados para mantener el prestigio de la Agencia.
3. En este sentido, la EMA promueve un acercamiento formalista a la transparencia, que se basa en la declaración de intereses de los partícipes en la evaluación de un medicamento, y que —por lo tanto— parte de la premisa de la voluntad subjetiva del sujeto afectado por el interés. Hay que encontrar fórmulas y criterios objetivos que puedan permitir baremar de forma independiente estas gravosas situaciones, que se manifiestan en las decisiones de comercialización.
4. El componente político está presente en la composición de la Agencia, sus comités, su consejo de administración, están formados por representantes de los Estados miembros: debería irse a un escenario donde prevalezcan mayoritariamente criterios objetivos, de especialización científica de sus componentes, eludiendo perspectivas de dependencia gubernamental, que sólo pueden ir en detrimento de la actividad de la EMA.
5. El gran punto de referencia para la Agencia es la FDA estadounidense, de cuya comparación se pueden sacar varias conclusiones: transparencia, celeridad en los procedimientos de aprobación —especialmente en relación con fármacos para enfermedades sin tratamiento, o para genéricos— consideración de los conflictos de interés y su gestión, eliminación de parámetros de representación territorial —siendo un país federal EE.UU.—, separación entre asesoría científica y decisión final. Todos son elementos que pueden ser contemplados en eventuales propuestas de mejora de la EMA.
6. Respecto a la comunicación de los resultados de las evaluaciones suscitadas en el marco de una autorización de comercialización, la Agencia puede catalogar de clasificados documentos por múltiples motivos —política presupuestaria o económica de los Estados miembros, afectación del interés general de los mismos o de las Instituciones Europeas, seguridad, etc.—, sin embargo, puede publicar resultados que afecten a información relevante a efectos comerciales. Es necesario aclarar los principios de publicidad y de comunicación, para que no respondan a intereses espurios, poco transparentes o claramente dañinos para el promotor de un determinado fármaco.
7. Se podría incluir como criterio en la selección y nombramiento de asesores, expertos e investigadores que colaboren con la EMA el principio de integridad científica, que sublimase cualquier otro baremo o consideración, para que la independencia de la Agencia fuese cada vez más real y auténtica, prescindiendo progresivamente de otro tipo de consideraciones periféricas que no pueden contribuir a la reputación de la misma.
8. No resulta admisible que la publicidad de las minutas y de las actas de votación se publiquen sólo parcialmente, partiendo de un principio de redacción o contenido mínimo, que es absolutamente incompatible con la declaración de principios que lleva a cabo la Agencia en su estatuto, al proclamar que pretende un nivel de transparencia superior al que tienen otros organismos e instituciones europeos.
9. La participación de expertos, científicos, asesores o investigadores con tachas relativas a su imparcialidad en las distintas fases de asesoría científica de la EMA tiene que quedar excluida: no basta con que no puedan votar, o participar en las deliberaciones previas, ya que sus opiniones quedarán manifestadas en sus informes o trabajos, lo que supone un hecho de dudosa admisibilidad porque puede influenciar a los que tomen la decisión final.
10. De este modo, los derechos de buena administración reconocidos en la Carta de Derechos Fundamentales de la UE, los principios proclamados en los Códigos de Buena Conducta Administrativa —tanto de la Comisión Europea como de la EMA— pueden quedar reducidos a vacuas y fútiles declaraciones, en la medida en que no se reflejen en la práctica cotidiana y corriente de la Agencia, de sus agentes, empleados y colaboradores, sin excluir otras lesiones más importantes sobre derechos fundamentales de la ciudadanía europea y declarados en la Carta de Derechos Fundamentales de la Unión Europea.
11. Por todo ello, hemos propugnado en este trabajo una revisión de los procedimientos internos de la EMA, que mejoren su funcionamiento, aportan claridad, transparencia y eficiencia en su labor, para que la promoción del bien común como interés público superior e indisponible inspire de manera efectiva y eficaz todo el trabajo de esta importante Agencia europea, y repercuta positivamente en el bien jurídico subyacente, la salud pública y su efectiva realización.
*El autor es Profesor Contratado Doctor en la Facultad de Derecho de la UCM, donde imparte Derecho Constitucional y Derecho de la Unión Europea,. Correo-e: jccano@ucm.es
1 Artículo 4.2. k) los asuntos comunes de seguridad en materia de salud pública, en los aspectos definidos en el presente Tratado
2 Alonso García, Ricardo, Sistema Jurídico de la Unión Europea, Madrid 2012, págs. 104 y ss.
3 Artículo 6 del TFUE: La Unión dispondrá de competencia para llevar a cabo acciones con el fin de apoyar, coordinar o complementar la acción de los Estados miembros. Los ámbitos de estas acciones serán, en su finalidad europea: a) la protección y mejora de la salud humana;
4 REGLAMENTO (CE) No 726/2004 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 31 de marzo de 2004 por el que se establecen procedimientos comunitarios para la autorización y el control de los medicamentos de uso humano y veterinario y por el que se crea la Agencia Europea de Medicamentos, DOUE de 30 de abril de 2004, L 136/1, que
5 https://www.ema.europa.eu/en
6 https://www.ema.europa.eu/en/search/search?search_api_views_fulltext=legislation
7 Pi Llorens, M., El nuevo mapa de las agencias europeas del Espacio de Libertad, Seguridad y Justicia. Revista de Derecho Comunitario Europeo, 56, 2017, págs. 77-117
8 https://european-union.europa.eu/institutions-law-budget/institutions-and-bodies/institutions-and-bodies-profiles_es?page=1
9 Renaud Dehousse, Regulation by networks in the European Community: the role of European agencies, Journal of European Public Policy, 1997, 4:2, págs. 246-261
10 Domenico Scarlattilaan 6, 1083 HS Amsterdam, Netherlands
11 Reglamento 726/2004: Artículo 64 1. El Director Ejecutivo será nombrado por el Consejo de Administración, por un período de cinco años, sobre la base de una lista de candidatos propuesta por la Comisión, tras la publicación de una convocatoria de manifestaciones de interés en el Diario Oficial de la Unión Europea y en otras publicaciones. Antes del nombramiento, se invitará sin demora al candidato designado por el Consejo de Administración a realizar una declaración ante el Parlamento Europeo y a responder a las preguntas formuladas por los miembros de esta institución. Su mandato podrá renovarse una vez. El Consejo de Administración podrá destituir al Director Ejecutivo a propuesta de la Comisión.
12 R. Alonso García, Derecho Comunitario: Sistema Constitucional y Administrativo de la Comunidad Europea, Ed. Centro de Estudios Ramón Areces, 1994.
13 Fuentetaja Pastor, Jesús Angel, Derecho Administrativo Europeo, Navarra 2020
14 SEC(2001)340 de 20 de febrero de 2001, p. 4.
16 Comisión Europea (2005, 5)
17 R. Alonso García y P.A. Sáenz de Santamaría, El sistema europeo de fuentes, Madrid 2022, Fundación Coloquio Jurídico Europeo
18 Artículo 308 Cuando una acción de la Comunidad resulte necesaria para lograr, en el funcionamiento del mercado común, uno de los objetivos de la Comunidad, sin que el presente Tratado haya previsto los poderes de acción necesarios al respecto, el Consejo, por unanimidad, a propuesta de la Comisión y previa consulta al Parlamento Europeo, adoptará las disposiciones pertinentes. (DOUE 2002/C 325/01 Unión Europea — Versiones consolidadas del Tratado de la Unión Europea y del Tratado constitutivo de la Comunidad Europea (2002)) .
19 Flynn, Leo, ‘Article 308 TFEU’, en Manuel Kellerbauer, Marcus Klamert, and Jonathan Tomkin (eds), The EU Treaties and the Charter of Fundamental Rights: A Commentary (New York, 2019; online edn, Oxford Academic)
20 Artículo 175 (*) 1. El Consejo, con arreglo al procedimiento previsto en el artículo 251 y previa consulta al Comité Económico y Social y al Comité de las Regiones, decidirá las acciones que deba emprender la Comunidad para la realización de los objetivos fijados en el artículo 174. (DOUE 2002/C 325/01 Unión Europea — Versiones consolidadas del Tratado de la Unión Europea y del Tratado constitutivo de la Comunidad Europea (2002))
21 Artículo 251 1. Cuando en el presente Tratado, para la adopción de un acto, se haga referencia al presente artículo, se aplicará el procedimiento siguiente. 2. La Comisión presentará una propuesta al Parlamento Europeo y al Consejo. El Consejo, por mayoría cualificada, previo dictamen del Parlamento Europeo: —si aprobara todas las enmiendas contenidas en el dictamen del Parlamento Europeo, podrá adoptar el acto propuesto así modificado, —si el Parlamento Europeo no propusiera enmienda alguna, podrá adoptar el acto propuesto, —en los demás casos, adoptará una posición común y la transmitirá al Parlamento Europeo. El Consejo informará plenamente al Parlamento Europeo de los motivos que le hubieran conducido a adoptar su posición común. La Comisión informará plenamente sobre su posición al Parlamento Europeo. (DOUE 2002/C 325/01 Unión Europea – Versiones consolidadas del Tratado de la Unión Europea y del Tratado constitutivo de la Comunidad Europea (2002))
22 Artículo 57.1
23 Artículo 65.1. El Consejo de Administración estará integrado por un representante de cada Estado miembro, dos representantes de la Comisión y dos representantes del Parlamento Europeo.
24 Artículo 65.2. Los miembros del Consejo de Administración serán nombrados atendiendo a sus conocimientos pertinentes en materia de gestión y, si procede, a su experiencia en el ámbito de los medicamentos de uso humano o veterinario
25 Artículo 65.6: 6. Las decisiones del Consejo de Administración se adoptarán por mayoría de dos terceras partes de sus miembros.
26 Artículo 65.8. El Consejo de Administración podrá invitar a los presidentes de los comités científicos a asistir a sus reuniones, con voz, pero sin voto.
27 Hofer, Matthias P., et al. “Impact of scientific advice from the European Medicines Agency.” Nature Reviews Drug Discovery, vol. 14, no. 5, May 2015, pp. 302
28 Artículo 61.1 in fine
29 Artículo 61.6. Tanto los miembros de los comités como los expertos responsables de la evaluación de los medicamentos se basarán en la experiencia y en los recursos científicos con que cuenten los organismos nacionales de autorización de comercialización. Cada autoridad nacional competente supervisará la categoría científica y la independencia de la evaluación realizada y facilitará las actividades de los miembros de los comités y de los expertos que haya designado. Los Estados miembros se abstendrán de dar a los miembros de los comités y a los expertos cualquier instrucción incompatible con las funciones que les incumban y con las tareas y responsabilidades de la Agencia.
30 Artículo 61.7. Con ocasión de la preparación de los dictámenes, cada comité hará todo lo posible por llegar a un consenso científico. Si no lo consiguiera, el dictamen recogerá la posición de la mayoría de los miembros, así como las posiciones divergentes y la motivación de las mismas
31 3. La Agencia publicará inmediatamente el informe de evaluación del medicamento de uso humano establecido por el Comité de medicamentos de uso humano, con los motivos de su dictamen, previa supresión de cualquier información comercial de carácter confidencial.
32 En caso de petición de reexamen contra uno de sus dictámenes, el Comité de que se trate nombrará a un ponente y, en su caso, a un ponente adjunto diferentes de los que haya designado para el dictamen inicial. Este procedimiento de reexamen sólo podrá referirse a elementos del dictamen inicial previamente identificados por el solicitante y basarse únicamente en los datos científicos disponibles en el momento de la adopción del dictamen inicial por parte del Comité. El solicitante podrá solicitar que el Comité consulte a un grupo científico consultivo en relación con tal tipo de reexamen.
Artículo 61.2 in fine: En caso de petición de reexamen contra uno de sus dictámenes, el Comité de que se trate nombrará a un ponente y, en su caso, a un ponente adjunto diferentes de los que haya designado para el dictamen inicial. Este procedimiento de reexamen sólo podrá referirse a elementos del dictamen inicial previamente identificados por el solicitante y basarse únicamente en los datos científicos disponibles en el momento de la adopción del dictamen inicial por parte del Comité. El solicitante podrá solicitar que el Comité consulte a un grupo científico consultivo en relación con tal tipo de reexamen.
33 Bakker, E., Plueschke, K., Jonker, C.J., Kurz, X., Starokozhko, V. and Mol, P.G.M., Contribution of Real-World Evidence in European Medicines Agency’s Regulatory Decision Making. Clin Pharmacol Ther, 2023, pág y ss. 113
34 Artículo 81 1. Toda decisión de concesión, denegación, modificación, suspensión, retirada o revocación de una autorización de comercialización, adoptada con arreglo al presente Reglamento, indicará de manera precisa los motivos en los que se base. La decisión se notificará al interesado.
35 Artículo 63.2. Los miembros del Consejo de Administración, los miembros de los comités, los ponentes y los expertos deberán carecer de intereses económicos o de cualquier otro tipo en la industria farmacéutica que pudieran poner en duda su imparcialidad. Se comprometerán a actuar al servicio del interés público y con un espíritu de independencia y presentarán anualmente una declaración sobre sus intereses económicos. Todos los intereses indirectos que puedan estar relacionados con la industria farmacéutica deberán constar en un registro que llevará la Agencia y que será accesible al público, previa solicitud, en las oficinas de la Agencia
36 Artículo 75.- El personal de la Agencia estará sujeto a los reglamentos y reglamentaciones aplicables a los funcionarios y otros agentes de las Comunidades Europeas. Respecto de su personal, la Agencia ejercerá las atribuciones conferidas a la autoridad facultada para proceder a los nombramientos. El Consejo de Administración, de acuerdo con la Comisión, adoptará las disposiciones de aplicación que resulten necesarias.
37 EMEA/45110/2007 -Revisado el 11 de octubre de 2021
38 Artículo 6.3 del Reglamento de Procedimiento para el Comité de Medicamentos de uso humano: For the evaluation of new marketing authorizations, Type II variation applications involving a new indication and referral procedures the rapporteur is supported by a co-rapporteur, as agreed by the Committee. The involvement of the co-rapporteur in other major variations will be decided by the CHMP on a case-by-case basis. A co-rapporteur shall be appointed from amongst the members of the Committee or alternates and shall prepare a critique of the rapporteur’s report or prepare a separate full report at the discretion of the Committee. The Committee may also appoint (a) peer reviewer(s) from amongst the members or alternates. In addition, the Committee may at any time ask for a peer review of any critical issues by the appropriate Committee’s working party or scientific advisory group.
39 Artículo 8.3: The divergent positions and the names of the members expressing the divergent positions shall be mentioned in the opinion of the Committee, and where relevant, the minutes of the Committee. Members having divergent positions shall provide them in writing, stating clearly the reasons on which they are based. They will be appended to the opinion. The reasons for the divergent opinions shall be publicly available together with the documentation made publicly available in relation to the evaluation of applications.
40 Bertele’, Vittorio MD; Bassi, Luca Li MD, DPH. A Critique to the European Regulatory System. Journal of Ambulatory Care Management 27(2): p. 98-104, April 2004
41 Regnstrom, J., Koenig, F., Aronsson, B. et al. Factors associated with success of market authorisation applications for pharmaceutical drugs submitted to the European Medicines Agency. Eur J Clin Pharmacol 66, 39–48 (2010)
42 La version actual de la Directiva es la que consta en el texto consolidado con fecha de 26 de mayo de 2021: Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use; Official Journal L 311, 28/11/2001 P. 0067 - 0128
43 Cfr. Publicación oficial de la EMA: “The European regulatory system for medicines Bringing new safe and effective medicines to patients across the European Union” 2023
44 Zhang, Y. (2022) Limits of Law in the Multilevel System: Explaining the European Commission’s Toleration of Noncompliance Concerning Pharmaceutical Parallel Trade. JCMS: Journal of Common Market Studies, 60: 1001– 1018
45 https://www.ema.europa.eu/en/human-regulatory/post-uthorisation/pharmacovigilance/medical-literature-monitoring
46 https://www.ema.europa.eu/en/human-regulatory/research-development/compliance/good-clinical-practice
47 Se pueden poner algunos ejemplos de preguntas planteadas durante la asesoría científica: Are the patients to be included in a study sufficiently representative of the population for whom the medicine is intended? Are the planned measures to assess the benefits of a medicine valid and relevant? Is the proposed plan to analyse results appropriate? Does the study last long enough and include enough patients to provide the necessary data for the benefit-risk assessment? Is the medicine being compared with an appropriate alternative? Are the plans to follow the long-term safety of the product appropriately designed? (se mantiene la version original en inglés, al ser la que utiliza la Agencia).
48 https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-advice-protocol-assistance
49 Schrama, R. (2023), Expert network interaction in the European Medicines Agency. Regulation & Governance, 17: 491-511
51 En el año 2015 el 15 por cien de las empresas que solicitaron asistencia científica de la EMA recibieron al final del procedimiento de aprobación una opinión negativa de la misma, frente al 25 por cien de las empresas que optaron por no solicitar la asistencia científica de la Agencia.
53 https://www.ema.europa.eu/en/human-regulatory/research-development/compliance/good-clinical-practice
54 Karolinska Institutet, https://ki.se/en
55 https://www.cnrs.fr/
56 https://www.scimagoir.com/institution.php?idp=21869
57 https://www.scimagoir.com/institution.php?idp=21629
58 https://www.medsci.ox.ac.uk/study/medicine
59 https://www.ema.europa.eu/en/about-us/what-we-do/authorisation-medicines/how-ema-evaluates-medicines
60 CHMP: Agendas, minutes and highlights | European Medicines Agency (europa.eu)
63 SINGH, Narayan B.; BJÖRLING, Elin A. A review of EMA assessment period reporting for mood variables in substance use research: Expanding existing EMA guidelines. Addictive behaviors, 2019, vol. 94, p. 133-146.
64 Cfr. Artículo 126a de la Directiva 2001/83/EC del Parlamento Europeo y del Consejo de 6 de noviembre de 2001, relativa al Código comunitario de medicamentos para uso humano.
66 https://www.ema.europa.eu/en/human-regulatory/overview/pharmacovigilance-overview
67 COM/2001/0428 final - Diario Oficial n° 287 de 12/10/2001 p. 0001 - 0029
68 https://ec.europa.eu/info/about-european-commission/service-standards-and-principles/transparency_es
69 ACUERDO INTERINSTITUCIONAL de 20 de mayo de 2021 entre el Parlamento Europeo, el Consejo de la Unión Europea y la Comisión Europea sobre un Registro de transparencia obligatorio
70 Cfr. N. 27.
71 Artículo 80: Artículo 80 Con el fin de garantizar un nivel de transparencia adecuado, el Consejo de Administración, a propuesta del Director Ejecutivo y de acuerdo con la Comisión, adoptará normas relativas a la puesta a disposición del público de informaciones reglamentarias, científicas o técnicas, relativas a la autorización y al control de medicamentos, que no tengan carácter confidencial. El reglamento interno y los procedimientos de la Agencia, de sus comités y de sus grupos de trabajo estarán a disposición del público en la Agencia y en Internet
72 EMA/555647/2013, 17 de diciembre de 2013, Stakeholders and Communication: Principles for publication of agendas and minutes of EMA scientific committees.
73 Reglamento N.º 1049/2001 del Parlamento Europeo y del Consejo de 30 de mayo de 2001, relativo al acceso público de los documentos del Parlamento Europeo, del Consejo y de la Comisión, DOUE de 31.5.2001, L 145-43.
74 Considerando (2) La apertura permite garantizar una mayor participación de los ciudadanos en el proceso de toma de decisiones, así como una mayor legitimidad, eficacia y responsabilidad de la administración para con los ciudadanos en un sistema democrático. La apertura contribuye a reforzar los principios de democracia y respeto de los derechos fundamentales contemplados en el artículo 6 del Tratado UE y en la Carta de los Derechos Fundamentales de la Unión Europea.
75 3. Principles: 3. Commercially confidential information and personal data will be redacted in the published documents. However, the aim should be to keep redaction to a minimum.
76 8. The minutes should provide a good quality reflection of the main discussions and conclusions/outcomes.
77 Rules for the implementation of Regulation (EC) No. 1049/2001 on access to EMEA documents. EMEA/MB/203359/2006 Rev. 1 Adopted, 19 December 2006.
78 Al no constar la traducción en castellano del mismo, las citas se harán conforme a la traducción llevada a cabo por el autor
79 Article 3.- Exceptions 1. The Agency shall refuse access to a document where disclosure would undermine the protection of: a) the public interest as regards public security, defence and military matters, international relations, the financial, monetary or economic policy of the Community or a Member State; b) privacy and the integrity of the individual, in particular in accordance with Community legislation regarding the protection of personal data.
80 2. The Agency shall refuse access to a document where disclosure would undermine the protection of: a) commercial interests of a natural or legal person, including intellectual property, b) court proceedings and legal advice, c) the purpose of inspections, investigations and audits, unless there is an overriding public interest in disclosure.
81 3. Access to a document, produced or received and in possession of the Agency, which relates to a matter where the decision has not been taken, shall be refused if disclosure of the document would seriously undermine the decision-making process, unless there is an overriding public interest in disclosure
82 3. (in fine) Access to a document containing opinions for internal use as part of deliberations and preliminary consultations within the Agency shall be refused even after the decision has been taken if disclosure of the document would seriously undermine the Agency’s decision-making process, unless there is an overriding public interest in disclosure.
83 4. As regards third-party documents, the Agency shall consult the third party with a view to assessing whether an exception in paragraph 1 or 2 is applicable, unless it is clear that the document shall or shall not be disclosed.
84 5. A Member State may request the Agency not to disclose a document originating from that Member State without its prior agreement.
85 5. A Member State may request the Agency not to disclose a document originating from that Member State without its prior agreement.
86 Kim, D.: «Transparency Policies of the European Medicines Agency: Has the Paradigm Shifted?», Medical Law Review, Oxford University Press, Oxford, 2017, vol. 25, n.º 3, pp. 456 a 483
87 Article 1 - Scope 1. The scope of this Decision is to ensure the widest possible access to the documents the Agency produces or receives and has in its possession.
88 Artículo 42 Derecho de acceso a los documentos Todo ciudadano de la Unión o toda persona física o jurídica que resida o tenga su domicilio social en un Estado miembro tiene derecho a acceder a los documentos del Parlamento Europeo, del Consejo y de la Comisión.
89 REGLAMENTO INTERNO DE LA COMISIÓN [C(2000) 3614] (DOUE L 308 de 8.12.2000, p. 26)
90 Ámbito El Código vincula a todo el personal cubierto por el Estatuto aplicable a los funcionarios y otros agentes de las Comunidades Europeas (en lo sucesivo denominado «el Estatuto») y las demás disposiciones sobre las relaciones entre la Comisión y su personal aplicables a funcionarios y otros agentes de las Comunidades Europeas. Sin embargo, las personas empleadas con contratos de Derecho privado, los expertos en comisión de servicios de la función pública nacional, los becarios, etc., que trabajan para la Comisión deberán también guiarse por él en su trabajo diario.
91 3. INFORMACIÓN SOBRE LOS DERECHOS DE LAS PARTES INTERESADAS
92 4.3. Clasificación a) En lo que atañe a la confidencialidad, se requieren cierta cautela y experiencia a la hora de seleccionar la información y el material que vayan a protegerse y evaluar el grado de protección requerido. Resulta fundamental que el grado de protección se corresponda con la importancia que revista, desde el punto de vista de la seguridad, el elemento concreto de información y material que haya de protegerse. A fin de garantizar la adecuada difusión de la información, se adoptarán las medidas necesarias para evitar una clasificación superior o inferior a la requerida.
93 European Medicines Agency Code of Good Administrative Behaviour, EMA/264257/2013
94 The Code of Good administrative behavior applies to all Agency staff employed under the Staff Regulations and the Conditions of employment of other servants. It further applies to other persons working for the Agency such as national experts on secondment, trainees, persons employed under private law contracts (interims), visiting experts in any work they perform in the context of the Agency’s role and in their relations with the public. Hereinafter, these persons are collectively referred to as “staff”.
95 Macrina, Francis L. Scientific integrity: Text and cases in responsible conduct of research. John Wiley & Sons, 2014.
96 6. Impartiality and independence. The staff of the Agency shall be impartial and independent, and respect principles of scientific integrity. They shall abstain from any arbitrary action adversely affecting members of the public or the Agency’s stakeholders, as well as from any preferential treatment on any grounds whatsoever. The staff of the Agency shall not be guided by any outside influences of whatever kind, including political or national influences, or by personal interests. They shall abstain from being involved in the taking of a decision on a matter concerning their own interests, or those of their family, relatives, and/or friends. In performing their role within the context of the Agency’s work scientific independence shall be ensured.
97 8. Legitimate expectations and consistency. The staff of the Agency shall be consistent in their own administrative behavior as well as with the administrative action of the Agency. They shall follow the Agency’s normal administrative practices, unless there are legitimate grounds for departing from those practices in an individual case. They shall respect the legitimate and reasonable expectations that members of the public have in the light of how the Agency has acted in the past, taking into account where circumstances require evolution of the Agency’s position.
98 4. Proportionality When taking decisions, the staff of the Agency shall ensure that the measures taken are proportional to the aim pursued. They shall in particular avoid restricting the rights of the citizens or imposing charges on them, when those restrictions or charges are not in a reasonable relation with the purpose of the action pursued. When taking decisions, they shall strike a fair balance between the legitimate interests of private persons and the general public interest.
99 European Medicines Agency Code of Conduct, 16 de junio de 2016, EMA/385894/2012 rev.1
100 Artículo 63.2. Los miembros del Consejo de Administración, los miembros de los comités, los ponentes y los expertos deberán carecer de intereses económicos o de cualquier otro tipo en la industria farmacéutica que pudieran poner en duda su imparcialidad. Se comprometerán a actuar al servicio del interés público y con un espíritu de independencia y presentarán anualmente una declaración sobre sus intereses económicos. Todos los intereses indirectos que puedan estar relacionados con la industria farmacéutica deberán constar en un registro que llevará la Agencia y que será accesible al público, previa solicitud, en las oficinas de la Agencia.
101 REGLAMENTO No 31 (CEE) 11 (CEEA) por el que se establece el Estatuto de los funcionarios y el régimen aplicable a los otros agentes de la Comunidad Económica Europea y de la Comunidad Europea de la Energía Atómica (DO 45 de 14.6.1962, p. 1385)
102 Guarantees of independence Article 19 1. The names of the members and alternates of the Committee shall be made public. When each appointment is published, the professional qualifications of each member and alternate shall be specified. 2. The members of the Committee and alternates, members of working parties, scientific advisory groups and experts mentioned in various articles of the present Rules of Procedure, shall not have any direct interests in the pharmaceutical industry, which could affect their impartiality. They shall undertake to act in the public interest and in an independent manner, and shall make an annual declaration of their financial interests. All indirect interests, which could relate to the pharmaceutical industry shall be entered in a register held by the Agency which is accessible to the public, on request at the Agency’s office. In addition, the Declarations of Interest of the members and alternates of the Committee shall be made available on the Agency’s website. 3. Members of the Committee and alternates, members of working parties and scientific advisory groups (and experts attending these meetings) shall declare at the beginning of each meeting any specific interests, which could be considered to be prejudicial to their independence with respect to the points of the agenda. These declarations shall be made available to the public.
103 SOP/EMA/0019
104 Artículo 76 Los miembros del Consejo de Administración, los miembros de los comités contemplados en el apartado 1 del artículo 56, los expertos, los funcionarios y los demás agentes de la Agencia estarán obligados, incluso después de haber cesado en sus funciones, a no divulgar las informaciones que, por su propia naturaleza, se hallen amparadas por el secreto profesional.
105 Artículo 17 1. El funcionario se abstendrá de divulgar sin autorización cualquier información que haya recibido con ocasión de sus funciones, salvo que dicha información se haya hecho ya pública o sea de acceso público. 2. El funcionario seguirá estando sujeto a esta obligación tras el cese de sus funciones. A
106 Artículo 17 bis 1. Todo funcionario tendrá derecho a la libertad de expresión, en el debido respeto de los principios de lealtad e imparcialidad. 2. Sin perjuicio de lo dispuesto en los artículos 12 y 17, todo funcionario que se proponga publicar o hacer publicar, individualmente o en colaboración, cualquier escrito que se refiera a la actividad de la Unión lo notificará previamente a la autoridad facultada para proceder a los nombramientos.
107 European Medicines Agency policy on the handling of competing interests of scientific committees’ members and experts, 11 de Junio de 2020, EMA/MB/89351/2020, que sustituye a la Policy/0044, de 6 de octubre de 2016 (EMA/626261/2014, Rev. 1).
108 Procedural Guidance on inclusion of declared interests in the European Medicines Agency’s electronic declaration of interest’s form (for scientific committees’ members and experts). 24 de abril 2015, EMA/627294/2014, Rev. 1.
109 3.2.2.1 Direct interests: -Employment with a pharmaceutical company, the biotechnology sector or the medical device sector. -Consultancy to a pharmaceutical company, the biotechnology sector or the medical device sector. -Strategic advisory role for a pharmaceutical company, the biotechnology or the medical device sector. -Financial interests in a pharmaceutical company, the biotechnology sector or the medical device sector. -Involvement of the expert in the repurposing of a medicinal product.
110 4.1. Objectives of the policy. The main objective of the policy is to ensure that the scientific committees’ members and the experts participating in the Agency’s activities have no interests in the pharmaceutical industry, and that CAT members and alternates have no interests in the biotechnology sector and the medical device sector, which could affect their impartiality, as per the requirements of EU legislation. This has to be balanced with the need to secure the best (specialist) scientific expertise for the evaluation and surveillance of medicinal products for human and veterinary use. It is, therefore, of utmost importance to strive for the optimal balance between the cooling-off period for the declared interests versus maintaining the experts’ knowledge.
111 https://www.fda.gov/drugs
112 White Paper -Office of Generic Drugs, Global Generic Drug Affairs: Comparing FDA and EMA Decisions for Market Authorization of Generic Drug Applications covering 2017-2020
113 Tacrolimus, anagrelide, paclitaxel generic, pág. 4 del White Paper, cit. 89.
114 Kashoski M., Hanaizi Z., Yordanova, S., Veselý R., Bouygues C., Llinares J., Kweder, S.: A Comparison of EMA and FDA Decisions for New Drug Marketing Applications 2014-2016: Concordance, Discordance, and Why, en Clinical, Pharmacology & Therapeutics, Volume 107 number 1, January 2020, pag. 6.
115 Downing NS., Aminawung JA. Shah N. et al., Regulatory Review of novel therapeutics-comparison of three regulatory agencies. New England Journal of Medicine 2012, 366:2284-93.
116 https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2020
117 Roberts SA, Allen JD, Sigal EV. Despite criticism of the FDA review process, new cancer drugs reach patient sooner in the United Stated than in Europe, en Health AFF (Millwood) 2011; 30-1375-81
118 MacLean CH, Morton SC, Ofman JJ, Roth EA, Shekelle PG: How useful are published data from the Food and Drug Administration in meta-analisys? en J Clin Epidem 2003; 56:44—51
119 Gatzsche PC, Jorgensen AW: Opening up data at the European Medicines Agency, en BMJ 2011; 342:d2686.
120 Robert Temple, “Development of Drug Law, Regulations, and Guidance in the U.S., in Principles of Pharmacology, 1994, p. 1646. (demasiada implicación por parte de los empleados de la FDA en el procedimiento de desarrollo provocaría que la Agencia no pudiese ser suficientemente neutral y analítica cuando los datos resultantes le fuesen presentados formando parte de un NDA. (traducción del autor).
121 Ibid., p. 1647
122 En concreto, dentro del Título 21, Capítulo I, Subcapítulo A, parte 19.
123 Egilman, A., Kapczynski, A., McCarthy, M., Luxkaranayagam, A., Morten, C., Herder, M., Ross, J. (2021). Transparency of Regulatory Data across the European Medicines Agency, Health Canada, and US Food and Drug Administration. Journal of Law, Medicine & Ethics, 49(3), 456-485
124 Cfr. cit. 91
125 https://www.fda.gov/drugs/development-approval-process-drugs
126 Dinis-Oliveira, Ricardo Jorge. COVID-19 research: pandemic versus “paperdemic”, integrity, values and risks of the “speed science”. Forensic sciences research, 2020, vol. 5, no 2, p. 174-187.
127 https://ec.europa.eu/info/law/law-making-process/adopting-eu-law/implementing-and-delegated-acts/comitology_en#:~:text=Comitology%20refers%20to/20a%20set,say%20in%20the%20implementing%20acts.
128 La comitología se reserva respecto de los actos de ejecución y los actos delegados, es decir, aquellos actos jurídicos de carácter general que articulan, detallan y expresan la ejecución de Reglamentos genéricos, y que por su naturaleza necesitan de una ulterior precisión para poder dotarlos de efectividad: de este modo, la Comisión Europea, en el marco de los mismos, utiliza frecuentemente el recurso a grupos y comités de expertos, de difícil imputación jurídica y cuya actividad tiene una compleja rendición de cuentas; por esta razón, se ha establecido una línea de transparencia que imbuya su actividad, y pueda mejorar su funcionamiento —por ejemplo, a través del Registro de comités—: https://ec.europa.eu/info/law/law—making—process/adopting—eu—law/implementing—and—delegated—acts/comitology_en#comitology—register
129 Sentencia del Tribunal de Justicia (Sala Tercera) de 22 de junio de 2023, en los Asuntos Acumulados C-6/21 P y C-16/21 P, Alemania y Estonia/Pharma Mar y Comisión
130 COMUNICADO DE PRENSA n.º 108/23 Luxemburgo, 22 de junio de 2023
131 Vid. N. 131
132 REGLAMENTO (UE) No 182/2011 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 16 de febrero de 2011 por el que se establecen las normas y los principios generales relativos a las modalidades de control por parte de los Estados miembros del ejercicio de las competencias de ejecución por la Comisión
133 https://ec.europa.eu/transparency/comitology-register/screen/committees
134 En este sentido, citar el informe del think tank Europa Ciudadana, Hiperregulación en la Unión Europea (https://www.europaciudadana.org/wp-content/uploads/2013/06/Informe_FEC_Hiperregulacion_.pdf).,
135 COM(2010) 543 final: Smart Regulation in the European Union https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=COM:2010:0543:FIN:EN:PDF
136 Case C-175/18 P PTC Therapeutics International Ltd v European Medicines Agency (EMA), and C-178/18 P MSD Animal Health Innovation and Intervet International v European Medicines Agency (EMA)
137 Artículo 4. Excepciones: 2. Las instituciones denegarán el acceso a un documento cuya divulgación suponga un perjuicio para la protección de:
- los intereses comerciales de una persona física o jurídica, incluida la propiedad intelectual,
- los procedimientos judiciales y el asesoramiento jurídico,
- el objetivo de las actividades de inspección, investigación y auditoría,
salvo que su divulgación revista un interés público superior.
138 Esta cuestión también se suscitó en el Asunto T-235/15, que implicó a Pari Pharma GmbH y la EMA, en relación con la puesta a disposición de un tercer solicitante información aportada respecto de la solicitud de autorización de comercialización del medicamento Vantobra.
139 Principles for Publication of agendas and minutes of EMA scientific committees, EMA/555647/2013: 4. Application of the principles.- Pre-opinion in the framework of marketing authorization applications: (…) The minutes will reflect the high-level procedure outcome or milestone —e.g. adoption of list of questions.
140 4. Application of the principles: Opinion in the framework of marketing authorization applications: (…) In case re-examination has been requested, the outcome of such re-examination has to be awaited before publication of the names of individuals expressing divergent views can take place.
141 Boucaud-Maitre, Denis; Altman, Jean-Jacques. Do the EMA accelerated assessment procedure and the FDA priority review ensure a therapeutic added value? 2006–2015: a cohort study. European journal of clinical pharmacology, 2016, vol. 72, p. 1275-1281.
142 Pérez Jaraba, María Dolores. Principios y reglas: examen del debate entre R. Dworkin y HLA Hart. Revista Estudios Jurídicos. Segunda Época, 2010, no 10.
143 Artículo 48 Presunción de inocencia y derechos de la defensa 1. Todo acusado se presume inocente hasta que su culpabilidad haya sido legalmente declarada. 2. Se garantiza a todo acusado el respeto de los derechos de la defensa.
144 Sentencia del Tribunal General (Sala Sexta) de 28 de octubre de 2020.
145 Commission Implementing Decision C(2018) 4831 final of 17 July 2018 refusing marketing authorisation under Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency (OJ 2004 L 136, p. 1) for Aplidin — plitidepsin, a medicinal product for human use.